ClinicoPath Module Development Guide
Source:vignettes/module-development-jamovi.Rmd
module-development-jamovi.RmdClinicoPath Module Development Guide
This comprehensive guide provides instructions for developing and maintaining the ClinicoPath jamovi module for clinicopathological research analysis.
📝 Development Focus: This guide focuses specifically on ClinicoPath module development. Generic jamovi tutorial content and examples using non-ClinicoPath datasets have been preserved in commented sections for reference.
🎯 Target Audience: R developers, statistical analysts, and researchers working with jamovi module development for clinical and pathological data analysis.
Guide Overview
- Prerequisites & Quick Setup - Development environment setup
- Advanced Development Patterns - Production-ready patterns
- Data Flow Architecture - Understanding the module architecture
- Developer Tips & Production Patterns - Best practices and patterns
- Common Pitfalls & Solutions - Troubleshooting guide
- Production Deployment - Release and deployment
- File Architecture Deep Dives - Detailed file format guides
- Reference Materials - Additional resources and examples
- Legacy Code Examples - Historical implementations for reference
Prerequisites & Quick Setup
ClinicoPath Development Setup
Prerequisites (one-time setup):
- Use
R >= 4.5.0 - Install jamovi from https://www.jamovi.org/download.html
- Install
jmvtools. It is not on CRAN - it must be installed from the jamovi repository, along with itsnodedependency:
install.packages('node', repos = 'https://repo.jamovi.org')
install.packages('jmvtools', repos = c('https://repo.jamovi.org', 'https://cran.r-project.org'))Build and install the module:
- Fork and clone this repository: https://github.com/sbalci/ClinicoPathJamoviModule
- Navigate to the cloned directory in R
- Run
jmvtools::install()to build and install the module - This produces
ClinicoPath.jmoand installs to jamovi - The module follows R package structure with additional
jamovi/folder
Key Development Areas
Core Analysis Modules
-
Stage Migration Analysis:
R/stagemigration.b.R- Advanced TNM staging validation with bootstrap and cross-validation -
Survival Analysis:
R/survival.b.R- Comprehensive survival analysis with Kaplan-Meier and Cox regression -
Decision Analysis:
R/decisiongraph.b.R- Medical decision trees and Markov chain models - Descriptive Analysis: Cross tables, descriptive statistics, and data checking tools
- Statistical Plots: JJStatsPlot integration for publication-ready visualizations
Core Development Guide
This section covers the essential development workflows, patterns, and best practices for ClinicoPath module development.
🏗️ Advanced Development Patterns & Best Practices
Enhanced Module Distribution System
This project uses a sophisticated module distribution system with the following features:
# Use the enhanced update system
Rscript _updateModules_enhanced.R
# Configuration-driven development
# Edit updateModules_config.yaml for module settingsKey Features: - ✅ Automated
testing and validation - ✅ Backup and
rollback capabilities
- ✅ Multi-module distribution to specialized repos -
✅ Configuration management via YAML - ✅
Security validation and integrity checks
ClinicoPath Development Workflow
# ClinicoPath-specific development cycle
clinicopath_development <- function() {
# 1. Modify existing analysis or create new one
# For existing: Edit R/stagemigration.b.R, R/survival.b.R, etc.
# For new: jmvtools::addAnalysis(name='newanalysis', title='New Analysis')
# 2. Update implementation files
# R/newanalysis.b.R - Main R6 class with .init(), .run() methods
# jamovi/newanalysis.a.yaml - Analysis options and parameters
# jamovi/newanalysis.u.yaml - User interface layout
# jamovi/newanalysis.r.yaml - Results tables and plots
# 3. ClinicoPath enhanced build process
jmvtools::prepare() # Generate .h.R files from .yaml
devtools::document() # Update .Rd files
jmvtools::prepare() # Critical: run twice for complex modules like stagemigration
devtools::document() # Ensure all documentation is current
# 4. Test and validate
devtools::test() # Run unit tests if available
jmvtools::install() # Build .jmo and install to jamovi
# 5. Update module configurations and documentation
# Edit _updateModules_config.yaml if new files added
# Update DESCRIPTION version if needed
# Update NEWS.md for significant changes
# 6. Distribute to sub-modules (ClinicoPath-specific)
system("Rscript _updateModules.R") # Distribute to specialized repos
}🔄 ClinicoPath Data Flow Architecture
📋 Note on Diagrams: - Mermaid Source Files: All diagram source code is available in
vignettes/*.mmdfiles - For Rendering: Use mermaid.live, GitHub preview, or mermaid CLI - See:vignettes/MERMAID_DIAGRAMS_README.mdfor detailed rendering instructions
Overall Module Data Flow
The ClinicoPath module follows a sophisticated data flow pattern that integrates user input, analysis processing, and result presentation:
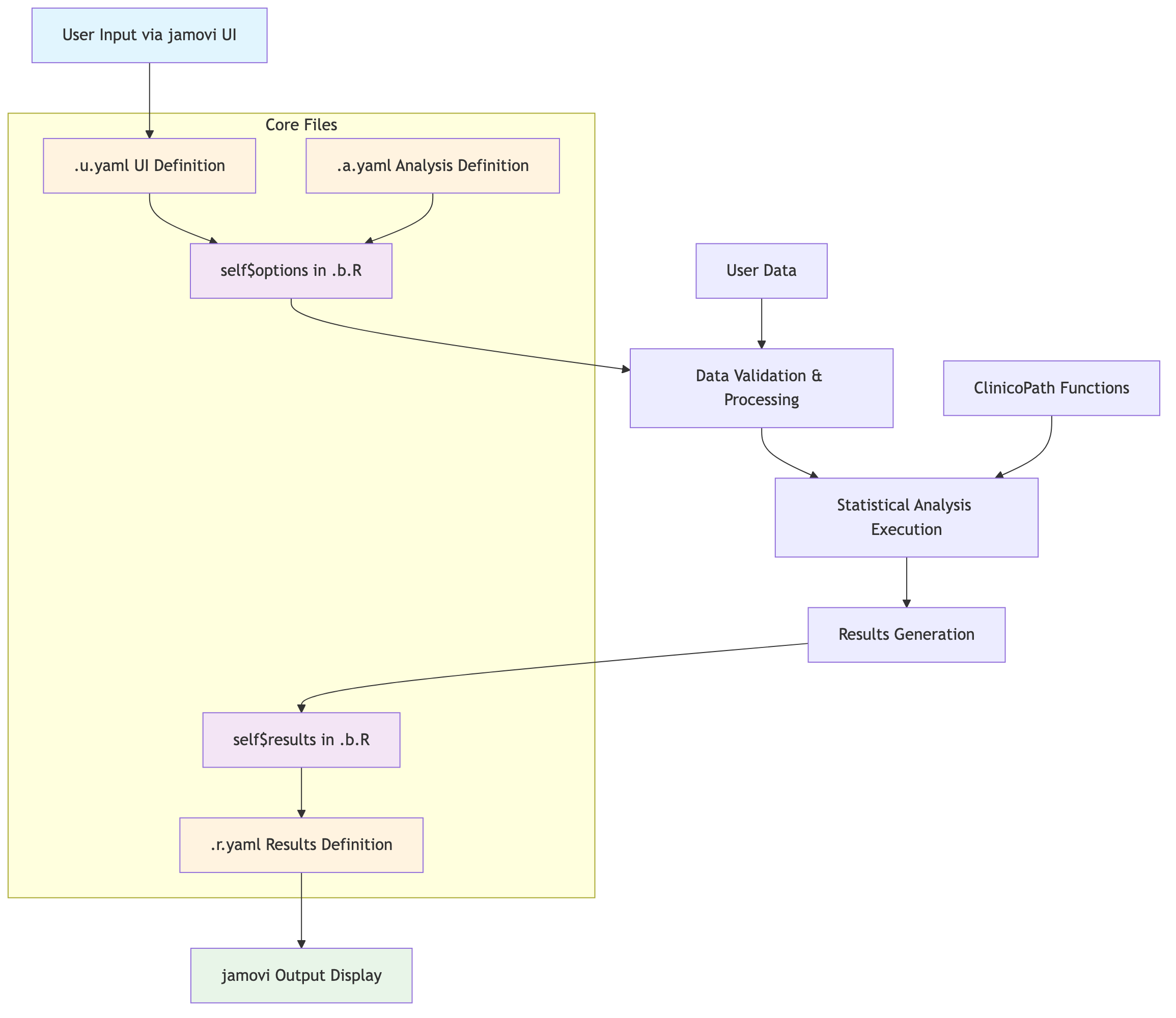
Source: vignettes/01-overall-data-flow.mmd
jamovi 4-File Architecture Interaction
The ClinicoPath module uses jamovi’s 4-file architecture with specific data flow between components:
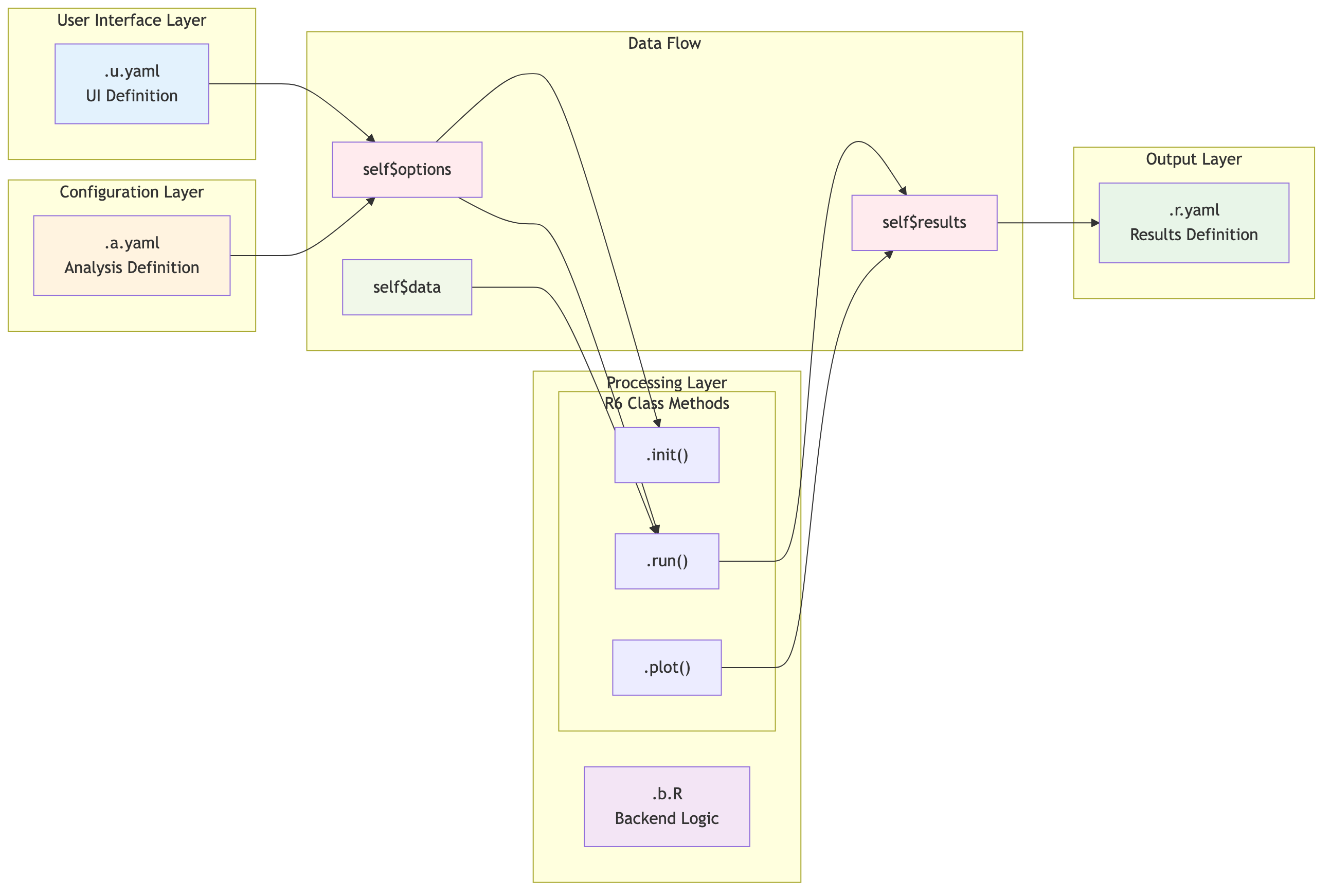
Source:
vignettes/02-jamovi-4file-architecture.mmd
Detailed Component Interaction Flow
This diagram shows how self$options and
self$results interact between the key files:
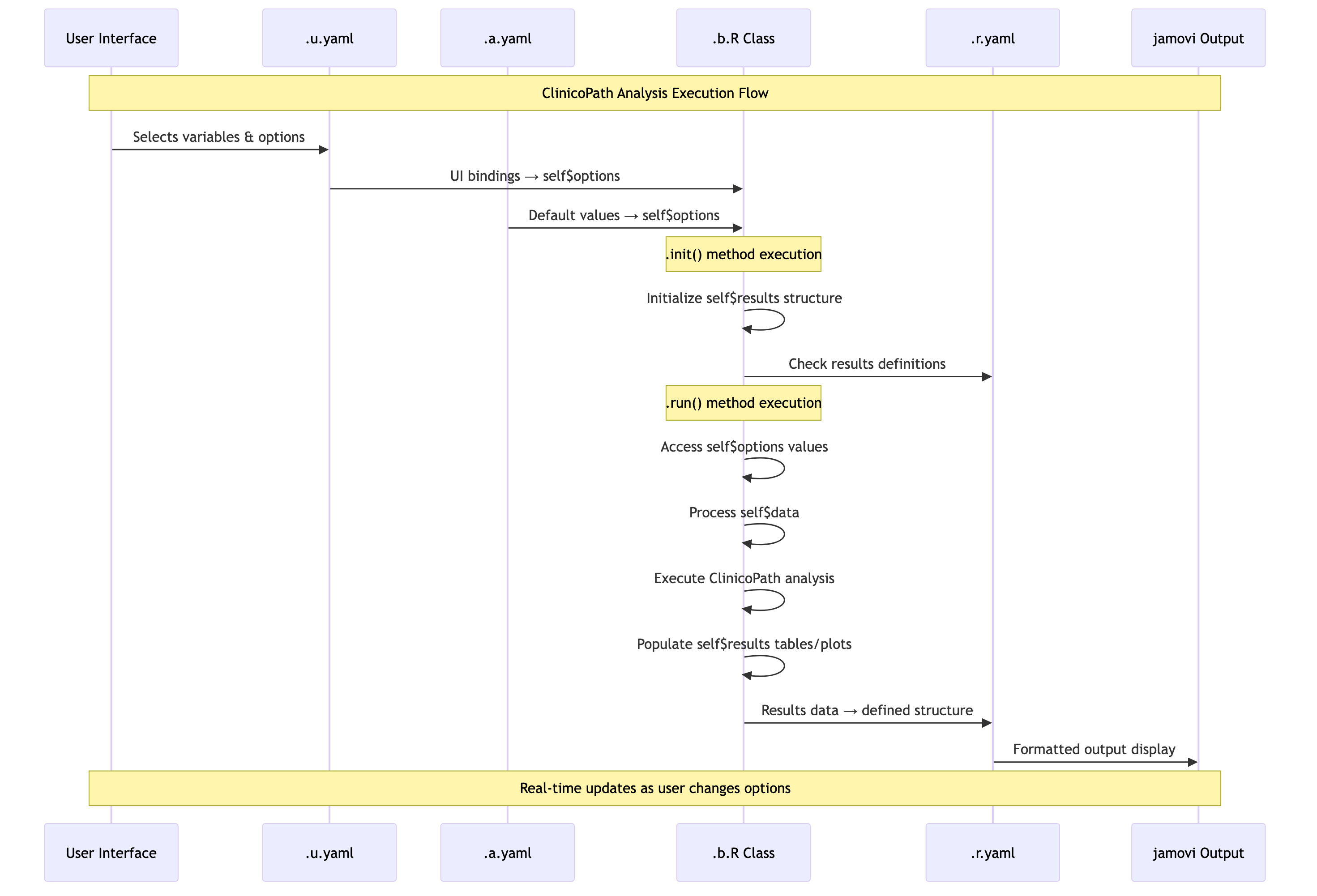
Source:
vignettes/03-component-interaction-sequence.mmd
ClinicoPath-Specific Data Processing Flow
For complex analyses like stagemigration, the data flow
includes multiple stages:
Simplified Text Flow:
Input Processing:
User Data → Data Validation → Variable Type Checking → Missing Data Handling
Analysis Configuration:
self$options$analysisType → {
- basic: Basic Migration Analysis
- standard: Standard + C-index Analysis
- comprehensive: Full Analysis Suite
- publication: Publication Ready Output
}
Core Analysis Engine:
Migration Matrix → Statistical Tests → C-index → Bootstrap → Cross-validation → Decision Curves
↘ Multivariable Analysis → Model Selection → Interaction Tests → Personalized Risk
Results Generation:
Migration Tables → Statistical Summaries → Diagnostic Plots → Clinical Interpretations → self$results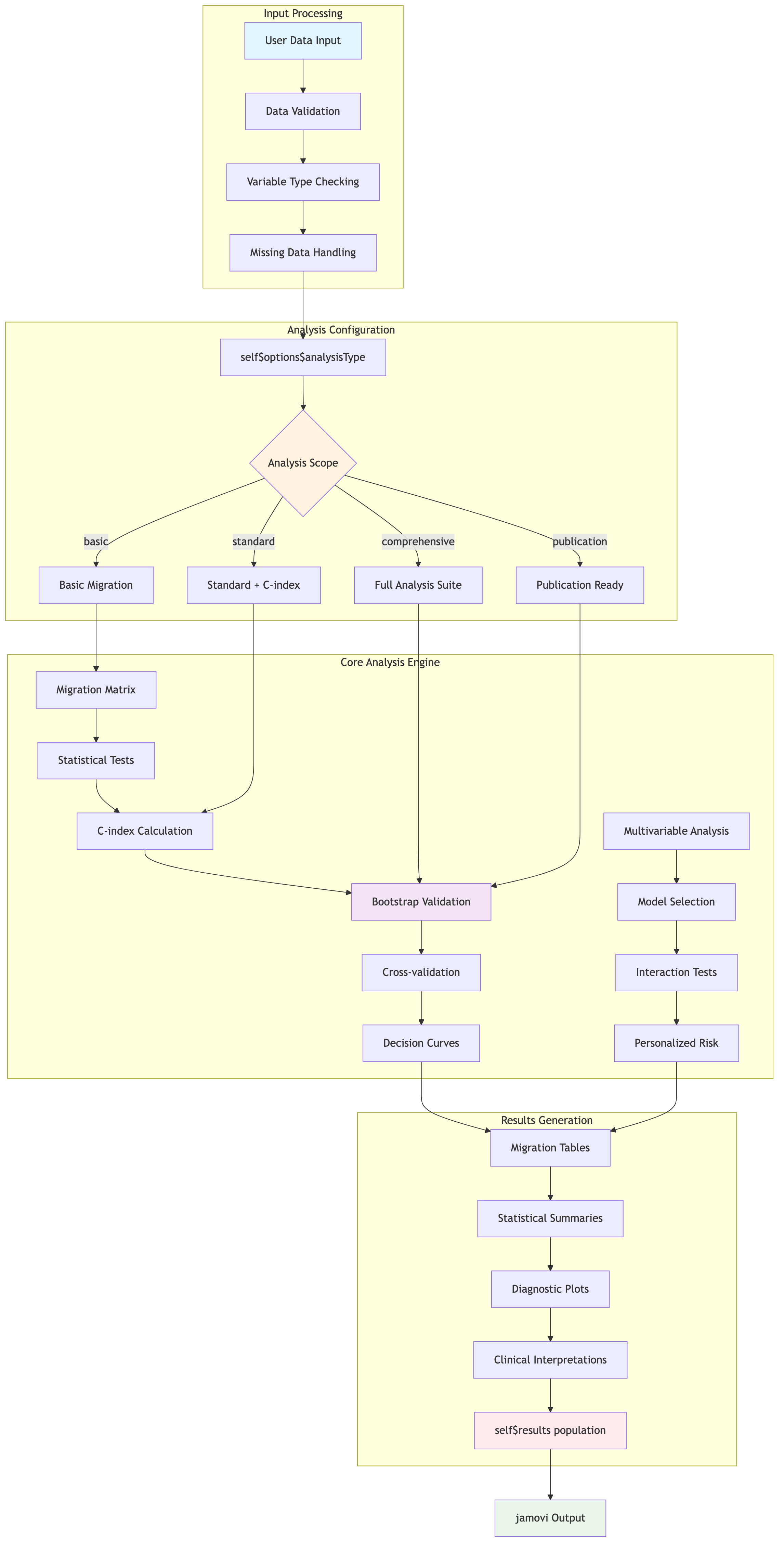
💡 Developer Tips & Production Patterns
1. Error Handling & User Experience
.run = function() {
# Always validate inputs first
if (is.null(self$options$dependent)) {
self$results$instructions$setContent(
"Please select a dependent variable to continue."
)
return()
}
# Graceful error handling
tryCatch({
# Your analysis code here
}, error = function(e) {
self$results$instructions$setContent(
paste("Analysis failed:", e$message)
)
return()
})
}4. Advanced Configuration Management
The project uses updateModules_config.yaml for
sophisticated module management:
5. Debugging Techniques
# 1. Use browser() for interactive debugging
.run = function() {
browser() # Pauses execution for inspection
# Your code here
}
# 2. Log intermediate results
.run = function() {
message("Starting analysis with ", nrow(self$data), " observations")
# Analysis steps with logging
result1 <- step1(self$data)
message("Step 1 completed, result has ", nrow(result1), " rows")
}
# 3. Use conditional debugging
.run = function() {
if (self$options$debug_mode) {
# Detailed debugging information
str(self$data)
str(self$options)
}
}🚨 Common Pitfalls & Solutions
2. UI Not Updating
# Clear jamovi cache
# Close jamovi completely
# Reinstall module
jmvtools::install()4. Memory Management for Large Datasets
.run = function() {
# Process data in chunks
chunk_size <- 1000
results <- list()
for (i in seq(1, nrow(data), chunk_size)) {
chunk <- data[i:min(i + chunk_size - 1, nrow(data)), ]
results[[length(results) + 1]] <- process_chunk(chunk)
# Clean up memory
gc()
# Checkpoint for user interruption
private$.checkpoint()
}
}🎯 Production Deployment Workflow
Module Distribution Commands
# 1. Prepare for distribution
jmvtools::prepare()
devtools::document()
devtools::test()
# 2. Update module configurations
# Edit updateModules_config.yaml
# 3. Distribute to submodules
system("Rscript _updateModules_enhanced.R")
# 4. Commit changes
system("git add .")
system("git commit -m 'Update modules for release'")
system("git push")📚 Documentation Best Practices
When Adding New Data and Vignettes
Important: When generating new example data and
vignettes, always add them to the appropriate place in
updateModules_config.yaml:
modules:
mymodule:
data_files:
- "new_example_data.rda" # Add here
vignette_files:
- "new_comprehensive_guide.qmd" # Add here
test_files:
- "test-new-function.R" # Add hereThis ensures your new files are distributed to submodules during the build process.
Comprehensive Documentation Pattern
#' Advanced Statistical Analysis
#'
#' @description
#' Performs comprehensive statistical analysis with modern methods
#' and publication-ready output.
#'
#' @details
#' This function implements state-of-the-art statistical methods with
#' proper error handling and user-friendly output.
#'
#' @examples
#' \dontrun{
#' # Basic usage
#' result <- advanced_analysis(
#' data = mtcars,
#' dependent = "mpg",
#' covariates = c("hp", "wt")
#' )
#' }
#'
#' @export
advanced_analysis <- function(data, dependent, covariates, method = "standard") {
# Implementation here
}File Architecture Deep Dives
This section provides comprehensive guides for each of the four jamovi file types that make up an analysis module.
🏛️ .b.R Function Architecture Deep Dive
Understanding the jamovi R6 Class Structure
Every jamovi analysis is implemented as an R6 class in a
.b.R file that inherits from an auto-generated base
class.
ClinicoPath Component Interaction Flow
The following diagram shows how ClinicoPath analyses like
stagemigration flow from configuration to results:
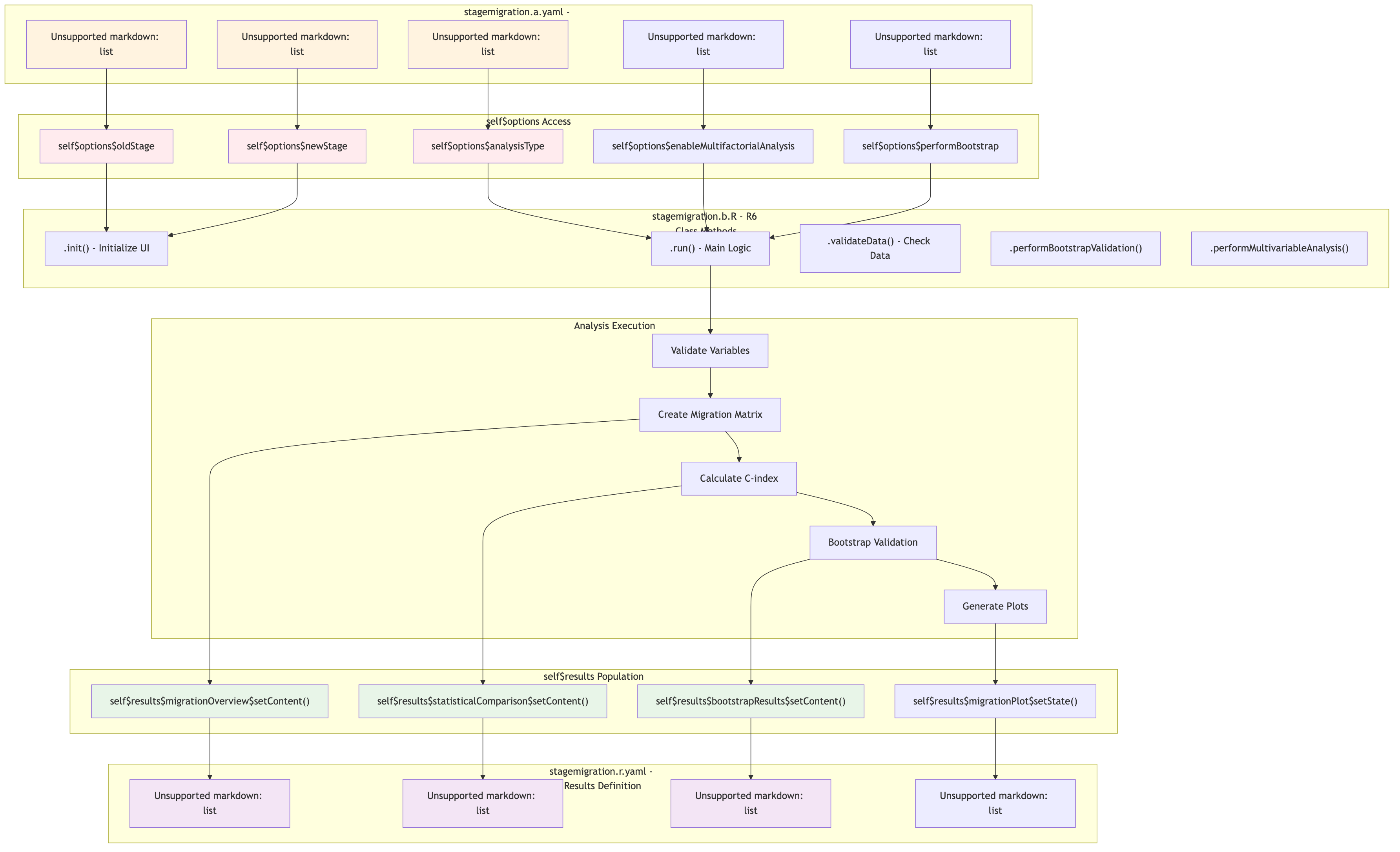
Source:
vignettes/05-stagemigration-component-flow.mmd
Detailed selfresults Interaction
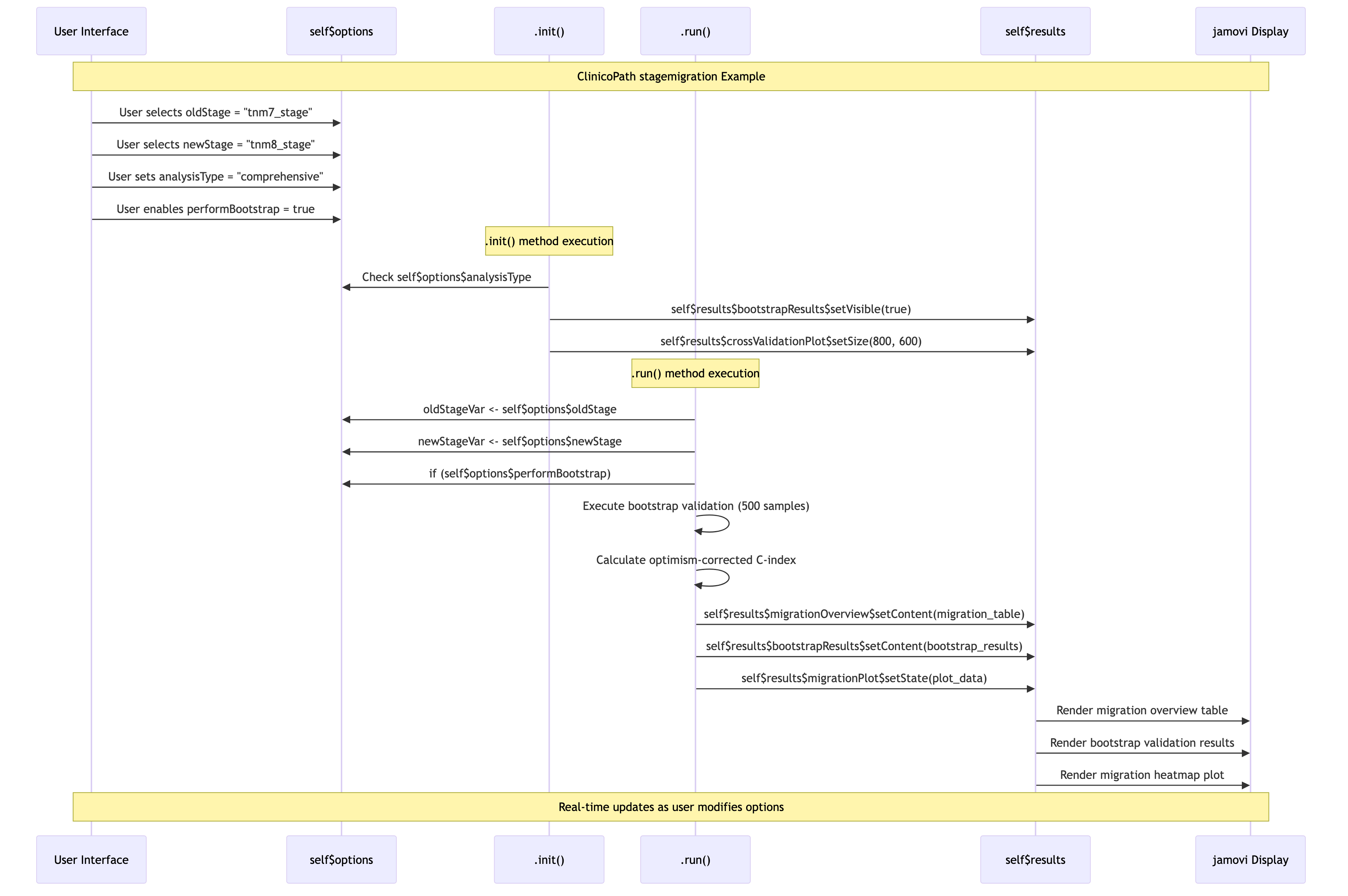
Source:
vignettes/06-stagemigration-detailed-interaction.mmd
Here’s the complete anatomy:
# Standard jamovi analysis structure
myanalysisClass <- if (requireNamespace('jmvcore'))
R6::R6Class(
"myanalysisClass",
inherit = myanalysisBase, # Auto-generated from .yaml files
private = list(
.init = function() { }, # Initialize UI and state
.run = function() { }, # Main analysis logic
.plot = function(image, ...) { }, # Plot generation
.getData = function() { }, # Data preprocessing (optional)
.labelData = function() { } # Label management (optional)
)
)Core Function Patterns & Lifecycle
1. .init() - Initialization & UI
Setup
This function runs when the analysis is first created or options change:
.init = function() {
# Dynamic UI sizing based on data
deplen <- length(self$options$dep)
self$results$plot$setSize(650, deplen * 450)
# Conditional visibility of results elements
if (self$options$advanced_options) {
self$results$advanced_table$setVisible(TRUE)
} else {
self$results$advanced_table$setVisible(FALSE)
}
# Data-driven UI updates
if (!is.null(self$options$grvar)) {
mydata <- self$data
grvar <- self$options$grvar
num_levels <- nlevels(as.factor(mydata[[grvar]]))
self$results$plot2$setSize(num_levels * 650, deplen * 450)
}
}Key Patterns: - 🎯 Dynamic sizing: Adjust plot dimensions based on data - 🔄 Conditional visibility: Show/hide UI elements based on options - 📊 Data-responsive UI: Adapt interface to data characteristics
2. .run() - Main Analysis Logic
The heart of every jamovi analysis with sophisticated error handling:
.run = function() {
# 1. Input Validation
if (is.null(self$options$dep) || is.null(self$options$group)) {
# User guidance with rich HTML
todo <- glue::glue("
<br>Welcome to ClinicoPath
<br><br>
This tool will help you generate [Analysis Name].
<br><br>
Please select:
<ul>
<li>Dependent variable</li>
<li>Grouping variable</li>
</ul>
<br>See documentation: <a href='...' target='_blank'>link</a>
<br><hr>
")
self$results$todo$setContent(todo)
return()
}
# 2. Data Validation
if (nrow(self$data) == 0) {
stop('Data contains no (complete) rows')
}
# 3. Progress Updates for Long Operations
self$results$todo$setContent("Processing analysis...")
# 4. Main Analysis with Checkpoints
for (i in seq_along(large_operation)) {
if (i %% 100 == 0) {
private$.checkpoint() # Allow user interruption
}
# Analysis steps...
}
# 5. Results Population
self$results$main_table$setContent(analysis_results)
self$results$plot$setState(plot_data)
}Key Patterns: - ✅ Progressive validation: Check inputs, then data, then proceed - 📝 Rich user guidance: HTML instructions with links and formatting - ⏱️ Checkpoint integration: Allow user interruption for long operations - 🔄 State management: Proper data flow to plots and tables
3. .plot() - Advanced Plot
Generation
Sophisticated plot generation with state management:
.plot = function(image, ggtheme, theme, ...) {
# 1. Validation
if (is.null(self$options$dep) || is.null(self$options$group)) {
return()
}
# 2. Retrieve plot data from state
plotData <- image$state
if (is.null(plotData)) {
return()
}
# 3. Theme handling
ggtheme <- private$.ggtheme()
theme_options <- private$.themeOptions()
# 4. Plot generation with error handling
tryCatch({
p <- ggplot2::ggplot(plotData, aes(x = x, y = y)) +
geom_point() +
ggtheme +
theme_options +
labs(
title = self$options$title,
subtitle = paste("n =", nrow(plotData)),
caption = "Generated by ClinicoPath"
)
# 5. Print and return
print(p)
TRUE
}, error = function(e) {
stop("Plot generation failed: ", e$message)
})
}4. .getData() - Data Preprocessing
Pipeline
Sophisticated data preprocessing with label management:
.getData = function() {
# 1. Get raw data
mydata <- self$data
mydata$row_names <- rownames(mydata)
# 2. Preserve original labels
original_names <- names(mydata)
labels <- setNames(original_names, original_names)
# 3. Clean variable names
mydata <- mydata %>% janitor::clean_names()
# 4. Restore labels after cleaning
corrected_labels <- setNames(original_names, names(mydata))
mydata <- labelled::set_variable_labels(
.data = mydata,
.labels = corrected_labels
)
# 5. Extract specific variables by label
all_labels <- labelled::var_label(mydata)
mytime <- names(all_labels)[all_labels == self$options$elapsedtime]
myoutcome <- names(all_labels)[all_labels == self$options$outcome]
# 6. Return structured data
return(list(
"mydata_labelled" = mydata,
"mytime_labelled" = mytime,
"myoutcome_labelled" = myoutcome
))
}Advanced Patterns & Best Practices
1. Label Management System
.labelData = function() {
mydata <- self$data
original_names <- names(mydata)
# Save original names as labels
labels <- setNames(original_names, original_names)
# Clean variable names
mydata <- mydata %>% janitor::clean_names()
# Apply original labels to cleaned names
cleaned_names <- names(mydata)
corrected_labels <- setNames(original_names, cleaned_names)
mydata <- labelled::set_variable_labels(
.data = mydata,
.labels = corrected_labels
)
return(mydata)
}2. Conditional Namespace Loading
All jamovi classes use conditional namespace loading for safety:
myanalysisClass <- if (requireNamespace('jmvcore'))
R6::R6Class(
# Class definition
)This prevents errors if core jamovi packages aren’t available.
3. Error Handling Patterns
# Pattern 1: Early return for missing inputs
if (is.null(self$options$required_var)) {
self$results$instructions$setContent("Please select required variable")
return()
}
# Pattern 2: Graceful error handling with user feedback
tryCatch({
analysis_result <- complex_analysis(data)
}, error = function(e) {
self$results$instructions$setContent(
paste("Analysis failed:", e$message,
"Please check your data and try again.")
)
return()
})
# Pattern 3: Data validation with specific messages
if (nrow(clean_data) < 10) {
self$results$instructions$setContent(
"Insufficient data. Need at least 10 complete observations."
)
return()
}4. State Management for Plots
# In .run() - prepare and transfer data to plot
plotData <- prepare_plot_data(analysis_results)
image <- self$results$plot
image$setState(plotData)
# In .plot() - retrieve and use the data
plotData <- image$state
if (is.null(plotData)) return()
# Generate plot using the transferred data
plot <- create_publication_plot(plotData)
print(plot)
TRUE5. Documentation Integration
#' @title Advanced Statistical Analysis
#' @description Comprehensive analysis with modern methods
#' @param data Data frame containing variables
#' @param dependent Character. Name of dependent variable
#' @param covariates Character vector. Covariate names
#' @details This function implements state-of-the-art methods...
#' @examples
#' \dontrun{
#' result <- myanalysis(data = mtcars, dependent = "mpg")
#' }
#' @importFrom R6 R6Class
#' @import jmvcore
#' @importFrom package function_namePerformance Optimization Patterns
1. Chunked Processing
.run = function() {
large_data <- self$data
chunk_size <- 1000
results <- list()
for (i in seq(1, nrow(large_data), chunk_size)) {
chunk_end <- min(i + chunk_size - 1, nrow(large_data))
chunk <- large_data[i:chunk_end, ]
# Process chunk
chunk_result <- process_chunk(chunk)
results[[length(results) + 1]] <- chunk_result
# Checkpoint for user interruption
private$.checkpoint()
# Memory cleanup
gc()
}
# Combine results
final_result <- do.call(rbind, results)
}2. Lazy Evaluation
.run = function() {
# Only compute expensive operations when needed
if (self$options$include_advanced) {
advanced_results <- expensive_computation(self$data)
self$results$advanced_table$setContent(advanced_results)
}
# Basic results are always computed
basic_results <- basic_analysis(self$data)
self$results$basic_table$setContent(basic_results)
}Integration with Enhanced Module System
When developing .b.R functions for the ClinicoPath ecosystem:
# Always follow the enhanced workflow
development_workflow <- function() {
# 1. Implement .b.R with proper structure
edit_analysis_file("R/myanalysis.b.R")
# 2. Update configuration
update_config_yaml("updateModules_config.yaml")
# 3. Enhanced build process
jmvtools::prepare()
devtools::document()
jmvtools::prepare() # Critical: run twice
devtools::document()
# 4. Test and distribute
devtools::test()
jmvtools::install()
system("Rscript _updateModules_enhanced.R")
}📋 .a.yaml Analysis Definition: Complete Architecture Guide
Understanding the .a.yaml Structure
The .a.yaml file defines the analysis metadata, options,
and interface behavior. It’s the blueprint for your jamovi analysis
module. Here’s the complete anatomy:
---
name: myanalysis # Internal analysis ID (must match .b.R filename)
title: My Statistical Analysis # Display name in jamovi menu
menuGroup: Exploration # Main menu category
menuSubgroup: Advanced Stats # Optional submenu
menuSubtitle: 'Detailed descriptive text' # Optional menu tooltip
version: '0.0.3' # Analysis version
jas: '1.2' # jamovi analysis specification version
description:
main: | # Main description (markdown supported)
Performs comprehensive statistical analysis with modern methods.
This analysis provides publication-ready output with effect sizes.
R: # R-specific documentation
dontrun: false # Whether to skip in R CMD check
usage: | # R usage examples
# Example usage:
ClinicoPath::myanalysis(
data = mtcars,
dep = "mpg",
group = "cyl"
)
options: # Analysis parameters/inputs
# ... detailed below
...Core Components Deep Dive
1. Menu System Architecture
# Menu positioning and organization
menuGroup: ExplorationD # Main menu category
menuSubgroup: ClinicoPath Comparisons # Submenu grouping
menuSubtitle: 'Univariate Survival Analysis, Cox, Kaplan-Meier' # Tooltip
# Menu groups used in ClinicoPath:
# - ExplorationD: Descriptive statistics
# - SurvivalD: Survival analysis
# - meddecideT: Medical decision making
# - JJStatsPlotD: Statistical visualizationsBest Practices: - Use descriptive
menuGroup names ending with ‘D’ for consistency - Keep
menuSubgroup concise but informative - Use
menuSubtitle for detailed descriptions that appear on
hover
2. Option Types & Patterns
Variable Selection Options
# Single variable selection
- name: dependent
title: Dependent Variable
type: Variable
suggested: [ continuous ] # Guides users to appropriate types
permitted: [ numeric ] # Enforces type restrictions
description: >
The outcome variable for analysis.
# Multiple variable selection
- name: covariates
title: Covariates
type: Variables # Note: Variables (plural) for multiple
suggested: [ ordinal, nominal, continuous ]
permitted: [ factor, numeric ]
default: NULL
# Variable with level selection
- name: outcome
title: Outcome Variable
type: Variable
suggested: [ ordinal, nominal ]
permitted: [ factor ]
- name: outcomeLevel
title: Event Level
type: Level
variable: (outcome) # Links to parent variable
description: >
Select which level represents the event of interest.
# Optional level selection
- name: referenceLevel
title: Reference Level (Optional)
type: Level
variable: (groupVar)
allowNone: true # Makes selection optionalList/Dropdown Options
- name: method
title: Analysis Method
type: List
options:
- title: Parametric (t-test)
name: parametric
- title: Non-parametric (Mann-Whitney U)
name: nonparametric
- title: Robust (Yuen's test)
name: robust
- title: Bayesian
name: bayes
default: parametric
description: >
Statistical method for group comparisons.Numeric Input Options
# Integer input
- name: bootstrapSamples
title: Bootstrap Samples
type: Integer
default: 1000
min: 100
max: 10000
# Decimal number input
- name: confidenceLevel
title: Confidence Level
type: Number
default: 0.95
min: 0.00
max: 1.00
description: >
Confidence level for intervals (e.g., 0.95 for 95% CI).String Input Options
- name: customTitle
title: Plot Title (Optional)
type: String
default: ''
description: >
Custom title for the plot. Leave empty for automatic title.
# String with specific format
- name: cutpoints
title: Time Cutpoints
type: String
default: '12, 36, 60'
description: >
Comma-separated time points for survival analysis (e.g., "12, 36, 60").Advanced Pattern: Conditional Options
# Parent option that controls visibility
- name: useAdvanced
title: Advanced Options
type: Bool
default: false
# Child option only visible when parent is true
- name: advancedMethod
title: Advanced Method
type: List
options:
- title: Method A
name: methodA
- title: Method B
name: methodB
default: methodA
visible: (useAdvanced) # Conditional visibility3. Complex Real-World Examples
Example 1: Survival Analysis Options
options:
# Time calculation options
- name: tint
title: Using Dates to Calculate Survival Time
type: Bool
default: false
description:
R: >
Calculate survival time from date variables.
- name: dxdate
title: Diagnosis Date
type: Variable
visible: (tint) # Only show when tint is true
description: >
Date of initial diagnosis.
- name: fudate
title: Follow-up Date
type: Variable
visible: (tint)
description: >
Most recent follow-up date.
# Analysis type with complex dependencies
- name: analysistype
title: Survival Type
type: List
options:
- title: Overall Survival
name: overall
- title: Cause Specific
name: cause
- title: Competing Risk
name: compete
default: overall
# Conditional level selections based on analysis type
- name: dod
title: Dead of Disease
type: Level
variable: (outcome)
allowNone: true
visible: (analysistype:cause || analysistype:compete)
- name: dooc
title: Dead of Other Causes
type: Level
variable: (outcome)
allowNone: true
visible: (analysistype:compete)Example 2: Statistical Test Configuration
options:
# Main test selection
- name: testType
title: Statistical Test
type: List
options:
- title: Automatic Selection
name: auto
- title: Chi-square Test
name: chisq
- title: Fisher's Exact Test
name: fisher
- title: Custom Test
name: custom
default: auto
# Conditional options for custom test
- name: customTestName
title: Custom Test Function
type: String
visible: (testType:custom)
default: ''
# Pairwise comparisons
- name: pairwise
title: Pairwise Comparisons
type: Bool
default: false
- name: pairwiseMethod
title: Adjustment Method
type: List
visible: (pairwise)
options:
- title: Holm
name: holm
- title: Hochberg
name: hochberg
- title: Bonferroni
name: bonferroni
- title: FDR
name: fdr
- title: None
name: none
default: holm4. Best Practices & Tips
Variable Type Specifications
# Complete type system reference
suggested: [ continuous, ordinal, nominal, id ]
permitted: [ numeric, factor, character, logical ]
# Common patterns:
# Continuous outcomes
suggested: [ continuous ]
permitted: [ numeric ]
# Categorical predictors
suggested: [ ordinal, nominal ]
permitted: [ factor ]
# Mixed types (flexible)
suggested: [ ordinal, nominal, continuous ]
permitted: [ factor, numeric ]
# ID variables
suggested: [ id ]
permitted: [ character, factor ]Description Best Practices
# Good: Clear, actionable description
- name: excludeOutliers
title: Exclude Outliers
type: Bool
default: false
description: >
Remove observations more than 3 standard deviations from the mean.
This uses the median absolute deviation (MAD) method for robust
outlier detection.
# Bad: Vague description
- name: option1
title: Option 1
type: Bool
description: >
Enable option 1.Default Value Guidelines
# Always provide sensible defaults
- name: confidenceLevel
type: Number
default: 0.95 # Standard 95% CI
- name: method
type: List
default: auto # Let analysis choose
- name: missing
type: List
options:
- title: Exclude listwise
name: exclude
- title: Include as NA
name: include
default: exclude # Safe default5. Advanced Patterns & Tricks
Dynamic UI Updates
# Options that trigger UI updates in .init()
- name: groupingVar
title: Grouping Variable
type: Variable
description: >
Variable for stratified analysis.
# In .b.R .init():
# if (!is.null(self$options$groupingVar)) {
# levels <- levels(self$data[[self$options$groupingVar]])
# self$results$groupTables$setTitle(
# paste("Results by", self$options$groupingVar)
# )
# }Complex Validation Patterns
# Date format handling
- name: dateFormat
title: Date Format in Data
type: List
options:
- title: YYYY-MM-DD
name: ymd
- title: MM/DD/YYYY
name: mdy
- title: DD/MM/YYYY
name: dmy
default: ymd
# Time unit conversion
- name: timeUnit
title: Time Unit
type: List
options:
- title: Days
name: days
- title: Weeks
name: weeks
- title: Months
name: months
- title: Years
name: years
default: monthsPerformance Optimization Options
# Computation control
- name: enableBootstrap
title: Bootstrap Confidence Intervals
type: Bool
default: false
description: >
Enable bootstrap CIs (slower but more accurate for small samples).
- name: bootstrapSamples
title: Bootstrap Iterations
type: Integer
visible: (enableBootstrap)
default: 1000
min: 100
max: 10000
- name: parallelProcessing
title: Use Parallel Processing
type: Bool
default: false
visible: (enableBootstrap)6. Integration with Module System
When creating .a.yaml files for the ClinicoPath system:
# 1. Follow naming conventions
name: myanalysis # Must match R/myanalysis.b.R
# 2. Use consistent menu groups
menuGroup: ExplorationD # Or SurvivalD, meddecideT, JJStatsPlotD
# 3. Version alignment
version: '0.0.3' # Match module version
# 4. Complete documentation
description:
main: |
Comprehensive description with markdown support.
- Feature 1: Description
- Feature 2: Description
R:
usage: |
# Complete R example
result <- ClinicoPath::myanalysis(
data = histopathology,
dependent = "outcome",
covariates = c("age", "stage"),
method = "robust"
)7. Common Pitfalls & Solutions
8. Testing Your .a.yaml
# Validate yaml syntax
yaml::yaml.load_file("jamovi/myanalysis.a.yaml")
# Check in jamovi
jmvtools::install()
# Open jamovi and verify:
# 1. Analysis appears in correct menu
# 2. All options are visible
# 3. Conditional visibility works
# 4. Defaults are sensibleReference Materials
This section contains additional resources, documentation links, and reference materials for jamovi module development.
📞 Getting Help & Resources
Development Resources
- Official Documentation: https://dev.jamovi.org/
- jamovi Community Forum: https://forum.jamovi.org/
- R Package Development: https://r-pkgs.org/
- Project Repository: Check GitHub issues and discussions
preparing development tools
use an unsigned version of jamovi for development in mac
jmvtools should be installed from the jamovi repo
https://dev.jamovi.org/tuts0101-getting-started.html
jmvtools is not on CRAN, so install it (and its
node dependency) from the jamovi repository:
install.packages('node', repos='https://repo.jamovi.org')
install.packages('jmvtools', repos=c('https://repo.jamovi.org', 'https://cran.r-project.org'))
jmvtools::check()
jmvtools::install()You can use devtools::install() to use your codes as a
usual R package, submit to github or CRAN.
devtools::check() does not like some jamovi folders so be
sure to add them under .Rbuildignore
Creating a Module
https://dev.jamovi.org/tuts0102-creating-a-module.html
jmvtools::create(path = "~/ClinicoPathDescriptives")Structure
Development Files
DESCRIPTION file
Imports, Depends, Suggests,
and Remotes have practically no difference in building
jamovi modules. The jmvtools::install() copies libraries
under build folder.
Under Imports jmvcore and R6
are defaults.
With Remotes one can install github packages as well. But with each
jmvtools::install() command it tries to check the updates,
and if you are online throws an error. An
upgrade = FALSE, quick = TRUE argument like in
devtools::install() is not available, yet. One workaround is temporarily
deleting Remotes from DESCRIPTION. The package folders continue to
remain under build folder.
One can also directly copy package folders from system R package
folder (find via .libPaths()) as well.
R folder
R folder is where the codes are present. There are two files.
function.b.R
Prepare Data For Analysis
varsName <- self$options$vars
data <- jmvcore::select(self$data, c(varsName))Remove NA containing cases (works on selected variables)
data <- jmvcore::naOmit(data)jmvcore::toNumeric() https://dev.jamovi.org/tuts0202-handling-data.html
can I just send whole data to plot function? you usually don’t want to, but sometimes it’s appropriate. normally you just provide a summary of the data to the plot function … just enough data for it to do it’s job. but if you need the whole data set for the plot function, then you can specify requiresData: true on the image object. that means the plot function can access self$data. i do it in the correlation matrix for example. there’s no summary i could send … the plot function needs all the data: https://github.com/jamovi/jmv/blob/master/jamovi/corrmatrix.r.yaml#L143 jamovi/corrmatrix.r.yaml:143 requiresData: true
jamovi folder
function.r.yaml
preformatted
Using “preformatted” result element I get a markdown table output. Is there a way to somehow render/convert this output to html version. Or should I go with https://dev.jamovi.org/api_table.html table api?
so you’re best to make use of the table api … the table API has a lot more features than an md table.
function.u.yaml
00refs.yaml
prepare a 00refs.yaml like this: https://github.com/jamovi/jmv/blob/master/jamovi/00refs.yaml
attach references to objects in the .r.yaml file like this:
https://github.com/jamovi/jmv/blob/master/jamovi/ancova.r.yaml#L174
Tables
I want a long table. I tried to use following but got error.
Below is my current .r.yaml - name: irrtable title: Interrater Reliability type: Table rows: 1 columns: - name: method title: ‘Method’ type: text - name: subjects title: ‘Subjects’ type: integer - name: raters title: ‘Raters’ type: integer - name: peragree title: ‘Agreement %’ type: number - name: kappa title: ‘Kappa’ type: number - name: z title: ‘z’ type: number - name: p title: ‘p-value’ type: number format: zto,pvalue
- try setting swapRowsColumns to true.
- alternatively, you can name your columns like this method[a], method[b], this will cause the row to be ‘folded’ where the value in method[b] appears below the value in method[a] . an example of this is the t-test: https://github.com/jamovi/jmv/blob/master/jamovi/ttestis.r.yaml#L20
.init()
so the principle seems right. you initialise the table in the .init() phase (you add rows and columns), and then you populate the table in the .run() phase. however, i notice your .init() function calls .initcTable() which doesn’t actually do anything.
most of the time, .init() isn’t necessary, because the .r.yaml file can take care of it, but sometimes the rows/columns the table should have is a more complex calculation than the .r.yaml allows (and example of this might be the ANOVA table in jmv … there’s not a simple relationship between the number of variables in the option, i.e. dose, supp, and the number of rows in the ANOVA table dose, supp, supp * dose, residuals. so we can’t achieve this with the .r.yaml, and so we set it up in the .init() phase.
finally, there are times where you can’t even determine the number of rows/columns in the .init() phase. you can only decide how many rows/columns are appropriate after you’ve run the analysis. an example of this might be a cluster analysis, where there’s a row for each cluster, but you only know how many rows you need after the analysis has been run. this is the least desireable, because it does lead to the growing and shrinking of the table, but sometimes that’s unavoidable.
so that’s your order of preference. preferably in the .r.yaml, if that can’t work, then do it in the .init(), and as a last resort, you can do it in the .run()Output variables in jamovi 1.6.16
hi, we’ve added “output variables” to version 1.6.16 of jamovi. this allows analyses to save data from the analyses, back to the spreadsheet (for example, residuals). there’s nothing in the 1.6.16 which indicates to users that this functionality is there, and it will only appear when an analysis implements these features. the idea is that we won’t actually release any modules with these features publicly, until an upcoming jamovi 1.8, or 2.0, or whatever. we’ve added these to the 1.6.16 so you can begin developing for the upcoming release.
you begin by specifying an output option in your .a.yaml file, i.e.
# - name: resids
# title: Residuals
# type: Output
# and then add an entry into your .r.yaml file, with a matching name:
# - name: resids
# title: Residuals
# type: Output
# varTitle: '`Residuals - ${ dep }`'
# varDescription: Residuals from ANCOVA
# clearWith:
# - dep
# - factors
# - covs
# - modelTerms
# in this case you’ll see that i’m specifying a formatted string, where the name of the column produced is generated from the dep variable, or dependent variable.
# you can populate the output column with:
# if (self$options$resids && self$results$resids$isNotFilled()) {
# self$results$resids$setValues(aVector)
# }
# sometimes your dataset will have gaps in it, either from filters, or from you calling na.omit() on it, and so if you simply send the residuals from your linear model to $setValues() they won’t be placed in the correct rows. there are two ways to solve this.
call self$results$resids$setRowNums(...) . conveniently, you can simply take the rownames() from your data set (after calling na.omit()) on it, and pass this in here. i.e.
# cleanData <- na.omit(self$data)
# ...
# rowNums <- rownames(cleanData)
# self$results$resids$setRowNums(rowNums)
# alternatively, you can turn your residuals into a data frame, attach the row numbers to that:
# residuals <- ...
# residuals <- data.frame(residuals=residuals, row.names=rownames(cleanData))
# self$results$setValues(residuals)
# if you want to provide multiple output columns, for example, perhaps in the previous example we want a “predicted values” column as well, we’d add additional entries to the .a.yaml and the .r.yaml. each entry in the .a.yaml will result in one checkbox.
# if you want to provide multiple columns with a single checkbox/option, then you can use the items property.
# - name: predInt
# title: Prediction intervals
# varTitle: Pred interval
# type: Output
# items: 2
# then you can go:
# self$results$predInt$setValues(index=i, values)
# or you could wrap both columns of values in a data frame, and go:
# self$results$predInt$setValues(valuesinadataframe)
# you can use data bindings with items too. i.e.
# - name: resids
# title: Residuals
# type: Output
# varTitle: 'Residuals - $key'
# items: (vars)
# this will create an output column for each variable assigned to vars. these can be set:
# self$results$resids$setValues(key=key, values)
Other Tips
Code Search in GitHub
https://github.com/search?l=&q=select+repo%3Ajamovi%2Fjmv+filename%3A.b.R+language%3AR&type=Code
select repo:jamovi/jmv filename:.b.R language:Rgenerate advanced search for all jamovi library
jamovi_library_names <- readLines("https://raw.githubusercontent.com/jonathon-love/jamovi-library/master/modules.yaml")
jamovi_library_names <- stringr::str_extract(
string = jamovi_library_names,
pattern = "github.com/(.*).git")
jamovi_library_names <- jamovi_library_names[!is.na(jamovi_library_names)]
jamovi_library_names <- gsub(pattern = "github.com/|.git",
replacement = "",
x = jamovi_library_names)
jamovi_library_names <- c("jamovi/jmv", jamovi_library_names)
jamovi_library_names <- gsub(pattern = "/",
replacement = "%2F",
x = jamovi_library_names)
query <- "type: Level"
repos <- paste0("repo%3A",jamovi_library_names,"+")
repos <- paste0(repos, collapse = "")
repos <- gsub(pattern = "\\+$",
replacement = "",
x = repos)
github_search <- paste0("https://github.com/search?q=",
query,
"+",
repos,
"&type=Code&ref=advsearch&l=&l=")
cat(github_search)Library Development Status
https://ci.appveyor.com/project/jonathon-love/jamovi-library/history
R version
Try to use compatible packages with the jamovi’s R version.
Use: R 4.5.0 https://cran.r-project.org/bin/macosx/r-release/R-4.5.0-x86_64.pkg
Use packages from CRAN snapshot:
options(
repos = "https://packagemanager.posit.co/cran/2025-05-25"
)Base R packages within jamovi
jamovi.app/Contents/Resources/modules/base/R
this folder contains base R packages used for jamovi.
jmvtools::install() prevent the packages already installed in base/R from being installed into your module.
(jmvtools is an R package which is a thin wrapper around the jamovi-compiler. The jamovi-compiler is written in javascript)
That cause problems if you are using different package versions. So it is best to keep up with suggested 'mran' version. Electron
jamovi is electron based. See R, shiny, and electron based application development here: Deploying a Shiny app as a desktop application with Electron
Project Structure
https://dev.jamovi.org/info_project-structure.html
https://forum.jamovi.org/viewtopic.php?f=12&t=1253&p=4251&hilit=npm#p4251
the easiest way to build jamovi on macOS is to use our dev bundle. https://www.jamovi.org/downloads/jamovi-dev.zip if you navigate to the
jamovi.app/Contents/Resourcesfolder, you’ll find a package.json which contains a bunch of different build commands. you can issue commands like: npm run build:client npm run build:server npm run build:analyses:jmv depending on which component you’re wanting to build.
Add Datasets
make a data folder (same as with an R package), and then you put entries in your 0000.yaml file:
https://github.com/gamlj/gamlj/blob/master/jamovi/0000.yaml#L47-L108
jamovi/0000.yaml:47-108
datasets:
- name: qsport
path: qsport.csv
description: Training hours
tags:.omv and .csv allowed. excel is also allowed but user does not see if it is csv or excel file.
Error messages
data <- data.frame(outcome=c(1,0,0,1,NA,1))
data <- na.omit(data)
if ( ! is.numeric(data$outcome) || any(data$outcome != 0 & data$outcome != 1))
stop('Outcome variable must only contains 1s and 0s')it’s good to test lots of different data sets that a user may have … include missing values, really large values, etc. etc. and make sure your analyses always handle them, and provide useful error messages for why an analysis doesn’t work. you don’t want to leave the user uncertain why something isn’t working … otherwise they just give up.
part of our philosophy is that people shouldn’t have to set their data up if they can’t be bothered … because with large data sets it can take a lot of time. so i’d encourage you to treat whatever the user provides you with as continuous, by converting it with toNumeric() … more on our data philosophy here: https://dev.jamovi.org/tuts0202-handling-data.html
in the options, you’ve got Survival Curve, and in the results, it’s Survival Plot … i’d encourage you to make these consistent. also, if the Survival Curve is unchecked, i’d hide the Surival plot, rather than leaving all that vacant space there.
visible: (optionName) https://github.com/jamovi/jmv/blob/master/jamovi/ttestis.r.yaml#L408-L416 jamovi/ttestis.r.yaml:408-416 - name: qq type: Image description: Q-Q plot width: 350 height: 300
is there a variable type for dates in jamovi? Can I force a user to add only a date to a VariablesListBox? I tried to get info from a selfvar via lubridate::is.Date and is.na.POSIXlt but it did not work hi, we don’t have a date data type at this time … only integer, numeric, and character … you could have people enter dates as character, and parse them yourself, but i appreciate that’s a bit of a hack
Thank you. Dates are always a problem in my routine practice. I work with many international colleagues and always date column is a mess, and people calculate survival time very differently. I want to have raw dates so that I can calculate survival time. I will try somehow going around.
learn YAML syntax
it’s a pretty straightforward syntax … you’ve basically got ‘objects’ where each of the elements have names, and you’ve got arrays, where each of the objects have an index. and that’s more-or-less all there is to it. you can take a look at jmv for examples: https://github.com/jamovi/jmv/tree/master/jamovi
I don’t think we’ve got a list of allowed parameters anywhere. Probably your best bet is to browse through the .yaml files in jmv. I think you’ll find there’s not that many parameter names.
as a work-around, once it’s installed the package from the Remotes, you can remove it from the DESCRIPTION and it won’t keep installing it over and over
Hi, there are scarce sources for pairwise chi-square tests. I have found rmngb::pairwise.chisq.test() and rmngb::pairwise.fisher.test() but that package has been removed from CRAN. Would you consider implementing this feature? I also thought to add these functions in a module, but I want to ask your policy about removed packages as well. 4 replies
jonathon:whale2: 18 days ago provided the module can be built with an entry in REMOTES, we don’t care if it’s not on CRAN
jonathon:whale2: 18 days ago … but you’re obviously taking a risk using something which isn’t maintained
Serdar Balci 18 days ago Thanks. Maybe just copying that function with appropriate reference may solve maintenance issue. I will think about it.
jonathon:whale2: 18 days ago oh yup
I have a question. I want to user to enter cut points in a box and then evaluate it as a vector. the function is this: summary(km_fit, times = c(12,36,60) I want user to define times vector. I have tried the following: utimes <- jmvcore::decomposeTerms(selfcutp) utimes <- as.vector(utimes) summary(km_fit, times = utimes a.yaml is as follows: - name: cutp title: Define at least two cutpoints (in months) for survival table type: String default: ‘12, 36, 60’ Would you please guide me to convert input into a vector. (edited) 3 replies
Serdar Balci 13 hours ago I think this seems to work: utimes <- selfcutp utimes <- strsplit(utimes, “,”) utimes <- purrr::reduce(utimes, as.vector) utimes <- as.numeric(utimes) (edited)
jonathon:whale2: 5 hours ago yup, this will do it too: as.numeric(strsplit(utimes, ‘,’)[[1]]) (it’s better if you can avoid using purrr, because it’s not really necessary, and you’re better off reducing the amount of dependencies you use)
Serdar Balci 5 hours ago thank you. :+1:
so wrt width/height, you can set that in the .r.yaml like so: https://github.com/kylehamilton/MAJOR/blob/master/jamovi/bayesmetacorr.r.yaml#L46-L49 it’s possible to do it programmatically, with … image$setSize()
Serdar Balci 4:48 PM
I think I am getting familiar with the codes :)
QuickTime Movie
JamoviModule.mov
4 MB QuickTime Movie - Click to downloadSerdar Balci Nov 29th, 2019 at 12:39 PM
Module names now have R version and OS in them. Does it mean that this will not work in windows Installing ClinicoPath_0.0.1-macos-R3.3.0.jmo
4 replies
jonathon:whale2: 3 months ago
It depends on whether there are any native R packages in your modules dependencies. Most modules do, but some don't. (You'll notice there's a "uses native" property there now too ... my intention is to use that to determine if a module can be used cross platform or not)
jonathon:whale2: 3 months ago
If there's native dependencies, then the module needs to be built separately for each os.
jonathon:whale2: 3 months ago
But I can take care of building it for different oses
Serdar Balci 3 months ago
Oh, I see. Thank you :slightly_smiling_face:Legacy Code Examples
⚠️ Historical Reference: The following sections contain legacy code examples, development notes, and historical implementations. This content is preserved for reference but may contain outdated patterns. Current development should follow the patterns described in the earlier sections of this guide.
Legacy Development Commands
library, eval=FALSE, include=FALSE
# install.packages('jmvtools', repos=c('https://repo.jamovi.org', 'https://cran.r-project.org'))
# jmvtools::check("C://Program Files//jamovi//bin")
# jmvtools::install(home = "C://Program Files//jamovi//bin")
#
# devtools::build(path = "C:\\ClinicoPathOutput")
# .libPaths(new = "C:\\ClinicoPathLibrary")
# devtools::build(path = "C:\\ClinicoPathOutput", binary = TRUE, args = c('--preclean'))
Sys.setenv(TZ="Europe/Istanbul")
library("jmvtools")
check, eval=FALSE, include=FALSE
jmvtools::check()
# rhub::check_on_macos()
# rhub::check_for_cran()
# codemetar::write_codemeta()
devtools::check()pkgdown build, eval=FALSE, include=FALSE
rmarkdown::render('/Users/serdarbalciold/histopathRprojects/ClinicoPath/README.Rmd', encoding = 'UTF-8', knit_root_dir = '~/histopathRprojects/ClinicoPath', quiet = TRUE)
devtools::document()
pkgdown::build_site()git force push, eval=FALSE, include=FALSE
# gitUpdateCommitPush
CommitMessage <- paste("updated on ", Sys.time(), sep = "")
wd <- getwd()
gitCommand <- paste("cd ", wd, " \n git add . \n git commit --message '", CommitMessage, "' \n git push origin master \n", sep = "")
# gitCommand <- paste("cd ", wd, " \n git add . \n git commit --no-verify --message '", CommitMessage, "' \n git push origin master \n", sep = "")
system(command = gitCommand, intern = TRUE)add analysis, eval=FALSE, include=FALSE
# jmvtools::install()
#
# jmvtools::create('ClinicoPath') # Use ClinicoPath instead of SuperAwesome
#
# jmvtools::addAnalysis(name='ttest', title='Independent Samples T-Test')
#
# jmvtools::addAnalysis(name='survival', title='survival')
#
# jmvtools::addAnalysis(name='correlation', title='correlation')
#
# jmvtools::addAnalysis(name='tableone', title='TableOne')
#
# jmvtools::addAnalysis(name='crosstable', title='CrossTable')
#
#
# jmvtools::addAnalysis(name='writesummary', title='WriteSummary')
# jmvtools::addAnalysis(name='finalfit', title='FinalFit')
# jmvtools::addAnalysis(name='multisurvival', title='FinalFit Multivariate Survival')
# jmvtools::addAnalysis(name='report', title='Report General Features')
# jmvtools::addAnalysis(name='frequencies', title='Frequencies')
# jmvtools::addAnalysis(name='statsplot', title='GGStatsPlot')
# jmvtools::addAnalysis(name='statsplot2', title='GGStatsPlot2')
# jmvtools::addAnalysis(name='scat2', title='scat2')
# jmvtools::addAnalysis(name='decisioncalculator', title='Decision Calculator')
# jmvtools::addAnalysis(name='agreement', title='Interrater Intrarater Reliability')
# jmvtools::addAnalysis(name='cluster', title='Cluster Analysis')
# jmvtools::addAnalysis(name='tree', title='Decision Tree')construct, eval=FALSE, include=FALSE
formula <- jmvcore::constructFormula(terms = c("A", "B", "C"), dep = "D")
jmvcore::constructFormula(terms = list("A", "B", c("C", "D")), dep = "E")
jmvcore::constructFormula(terms = list("A", "B", "C"))
vars <- jmvcore::decomposeFormula(formula = formula)
unlist(vars)
cformula <- jmvcore::composeTerm(components = formula)
jmvcore::composeTerm("A")
jmvcore::composeTerm(components = c("A", "B", "C"))
jmvcore::decomposeTerm(term = c("A", "B", "C"))
jmvcore::decomposeTerm(term = formula)
jmvcore::decomposeTerm(term = cformula)
composeTerm <- jmvcore::composeTerm(components = c("A", "B", "C"))
jmvcore::decomposeTerm(term = composeTerm)
Example
read data, eval=FALSE, include=FALSE
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))writesummary, eval=FALSE, include=FALSE
devtools::install(upgrade = FALSE, quick = TRUE)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
# library("ClinicoPath")
deneme$Age <- as.numeric(as.character(deneme$Age))
ClinicoPath::writesummary(data = deneme, vars = Age)
ggstatsplot::normality_message(deneme$Age, "Age")
ClinicoPath::writesummary(
data = deneme,
vars = Age)
finalfit, eval=FALSE, include=FALSE
devtools::install(upgrade = FALSE, quick = TRUE)
library(dplyr)
library(survival)
library(finalfit)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
ClinicoPath::finalfit(data = deneme,
explanatory = Sex,
outcome = Outcome,
overalltime = OverallTime)decision, eval=FALSE, include=FALSE
devtools::install(upgrade = FALSE, quick = TRUE)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
ClinicoPath::decision(
data = deneme,
gold = Outcome,
goldPositive = "1",
newtest = Smoker,
testPositive = "TRUE")
ClinicoPath::decision(
data = deneme,
gold = LVI,
goldPositive = "Present",
newtest = PNI,
testPositive = "Present")eval=FALSE, include=FALSE
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
ggstatsplot::ggbetweenstats(data = deneme,
x = LVI,
y = Age)statsplot, eval=FALSE, include=FALSE
devtools::install(upgrade = FALSE, quick = TRUE)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
ClinicoPath::statsplot(
data = deneme,
dep = Age,
group = Smoker)decision 2, eval=FALSE, include=FALSE
mytable <- table(deneme$Outcome, deneme$Smoker)
caret::confusionMatrix(mytable)
confusionMatrix(pred, truth)
confusionMatrix(xtab, prevalence = 0.25)
levels(deneme$Outcome)
mytable[1,2]
d <- "0"
mytable[d, "FALSE"]
mytable[[0]]construct formula, eval=FALSE, include=FALSE
formula <- jmvcore::constructFormula(terms = c("A", "B", "C"))
jmvcore::constructFormula(terms = list("A", "B", "C"))
vars <- jmvcore::decomposeFormula(formula = formula)
vars <- jmvcore::decomposeTerms(vars)
vars <- unlist(vars)
formula <- as.formula(formula)
my_group <- "lvi"
my_dep <- "age"
formula <- paste0('x = ', group, 'y = ', dep)
myformula <- as.formula(formula)
myformula <- glueformula::gf(my_group, my_dep)
myformula <- glue::glue( 'x = ' , my_group, ', y = ' , my_dep)
myformula <- jmvcore::composeTerm(myformula)
eval=FALSE, include=FALSE
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
library(survival)
km_fit <- survfit(Surv(OverallTime, Outcome) ~ LVI, data = deneme)
library(dplyr)
km_fit_median_df <- summary(km_fit)
km_fit_median_df <- as.data.frame(km_fit_median_df$table) %>%
janitor::clean_names(dat = ., case = "snake") %>%
tibble::rownames_to_column(.data = ., var = "LVI")construct formula 2, eval=FALSE, include=FALSE
library(dplyr)
library(survival)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
myoveralltime <- deneme$OverallTime
myoutcome <- deneme$Outcome
myexplanatory <- deneme$LVI
class(myoveralltime)
class(myoutcome)
typeof(myexplanatory)
is.ordered(myexplanatory)
formula2 <- jmvcore::constructFormula(terms = "myexplanatory")
# formula2 <- jmvcore::decomposeFormula(formula = formula2)
# formula2 <- paste("", formula2)
# formula2 <- as.formula(formula2)
formula2 <- jmvcore::composeTerm(formula2)
formulaL <- jmvcore::constructFormula(terms = "myoveralltime")
# formulaL <- jmvcore::decomposeFormula(formula = formulaL)
formulaR <- jmvcore::constructFormula(terms = "myoutcome")
# formulaR <- jmvcore::decomposeFormula(formula = formulaR)
formula <- paste("Surv(", formulaL, ",", formulaR, ")")
# formula <- jmvcore::composeTerm(formula)
# formula <- as.formula(formula)
# jmvcore::constructFormula(terms = c(formula, formula2))
deneme %>%
finalfit::finalfit(formula, formula2) -> tUni
tUnieval=FALSE, include=FALSE
library(dplyr)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
results <- deneme %>%
ggstatsplot::ggbetweenstats(LVI, Age)
results
mydep <- deneme$Age
mygroup <- deneme$LVI
mygroup <- jmvcore::constructFormula(terms = "mygroup")
mygroup <- jmvcore::composeTerm(mygroup)
mydep <- jmvcore::constructFormula(terms = "mydep")
mydep <- jmvcore::composeTerm(mydep)
# not working
# eval(mygroup)
# rlang::eval_tidy(mygroup)
# !!mygroup
# mygroup
# sym(mygroup)
# quote(mygroup)
# enexpr(mygroup)
mygroup <- jmvcore::constructFormula(terms = "mygroup")
mydep <- jmvcore::constructFormula(terms = "mydep")
formula1 <- paste(mydep)
formula1 <- jmvcore::composeTerm(formula1)
mygroup <- paste(mygroup)
mygroup <- jmvcore::composeTerm(mygroup)
mydep <- deneme$Age
mygroup <- deneme$LVI
mydep <- jmvcore::resolveQuo(jmvcore::enquo(mydep))
mygroup <- jmvcore::resolveQuo(jmvcore::enquo(mygroup))
mydata2 <- data.frame(mygroup=mygroup, mydep=mydep)
results <- mydata2 %>%
ggstatsplot::ggbetweenstats(
x = mygroup, y = mydep )
results
myformula <- glue::glue('x = ', mygroup, ', y = ' , mydep)
myformula <- jmvcore::composeTerm(myformula)
myformula <- as.formula(myformula)
mydep2 <- quote(mydep)
mygroup2 <- quote(mygroup)
results <- deneme %>%
ggstatsplot::ggbetweenstats(!!mygroup2, !!mydep2)
resultsconstruct formula 3, eval=FALSE, include=FALSE
formula <- jmvcore::constructFormula(terms = c("myoveralltime", "myoutcome"))
vars <- jmvcore::decomposeFormula(formula = formula)
explanatory <- jmvcore::constructFormula(terms = c("explanatory"))
explanatory <- jmvcore::decomposeFormula(formula = explanatory)
explanatory <- unlist(explanatory)
myformula <- paste("Surv(", vars[1], ", ", vars[2], ")")
deneme %>%
finalfit::finalfit(myformula, explanatory) -> tUnitable tangram, eval=FALSE, include=FALSE
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
table3 <-
tangram::html5(
tangram::tangram(
"Death ~ LVI + PNI + Age", deneme),
fragment=TRUE,
inline="nejm.css",
caption = "HTML5 Table NEJM Style",
id="tbl3")
table3
mydep <- deneme$Age
mygroup <- deneme$Death
formulaR <- jmvcore::constructFormula(terms = c("LVI", "PNI", "Age"))
formulaL <- jmvcore::constructFormula(terms = "Death")
formula <- paste(formulaL, '~', formulaR)
formula <- as.formula(formula)
table <- tangram::html5(
tangram::tangram(formula, deneme
))
table
arsenal
arsenal, results='asis', eval=FALSE, include=FALSE
tab1 <- arsenal::tableby(~ Age + Sex, data = deneme)
results <- summary(tab1)
# results$object
# results$control
# results$totals
# results$hasStrata
# results$text
# results$pfootnote
# results$term.name
#
# tab1$Call
#
# tab1$control
tab1$tables # this is where results lie
Multivariate Analysis Survival
Multivariate Analysis, eval=FALSE, include=FALSE
library(finalfit)
library(survival)
explanatoryMultivariate <- explanatoryKM
dependentMultivariate <- dependentKM
mydata %>%
finalfit(dependentMultivariate, explanatoryMultivariate) -> tMultivariate
knitr::kable(tMultivariate, row.names=FALSE, align=c("l", "l", "r", "r", "r", "r"))eval=FALSE, include=FALSE
# Find arguments in yaml
list_of_yaml <- c(
list.files(path = "~/histopathRprojects/ClinicoPath-Jamovi--prep/jmv",
pattern = "\\.yaml$",
full.names = TRUE,
all.files = TRUE,
include.dirs = TRUE,
recursive = TRUE
)
)
text_of_yaml_yml <- purrr::map(
.x = list_of_yaml,
.f = readLines
)
text_of_yaml_yml <- as.vector(unlist(text_of_yaml_yml))
arglist <-
stringr::str_extract(
string = text_of_yaml_yml,
pattern =
"([[:alnum:]]*):"
)
arglist <- arglist[!is.na(arglist)]
arglist <- unique(arglist)
arglist <- gsub(pattern = ":", # remove some characters
replacement = "",
x = arglist)
arglist <- trimws(arglist) # remove whitespace
cat(arglist, sep = "\n")
#
# # tUni_df_descr <- paste0("When ",
# # tUni_df$dependent_surv_overall_time_outcome[1],
# # " is ",
# # tUni_df$x[2],
# # ", there is ",
# # tUni_df$hr_univariable[2],
# # " times risk than ",
# # "when ",
# # tUni_df$dependent_surv_overall_time_outcome[1],
# # " is ",
# # tUni_df$x[1],
# # "."
# # )
#
# # results5 <- tUni_df_descreval=FALSE, include=FALSE
boot::melanoma
rio::export(x = boot::melanoma, file = "data/melanoma.csv")
survival::colon
rio::export(x = survival::colon, file = "data/colon.csv")
# BreastCancerData <- "https://archive.ics.uci.edu/ml/machine-learning-databases/breast-cancer-wisconsin/breast-cancer-wisconsin.data"
#
# BreastCancerNames <- "https://archive.ics.uci.edu/ml/machine-learning-databases/breast-cancer-wisconsin/breast-cancer-wisconsin.names"
#
# BreastCancerData <- read.csv(file = BreastCancerData, header = FALSE,
# col.names = c("id","CT", "UCSize", "UCShape", "MA", "SECS", "BN", "BC", "NN","M", "diagnosis") )
library(mlbench)
data("BreastCancer")
BreastCancer
rio::export(x = BreastCancer, file = "data/BreastCancer.csv")pairwise, eval=FALSE, include=FALSE
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
# names(deneme)
mypairwise <- survminer::pairwise_survdiff(
formula = survival::Surv(OverallTime, Outcome) ~ TStage,
data = deneme,
p.adjust.method = "BH"
)
mypairwise2 <- as.data.frame(mypairwise[["p.value"]]) %>%
tibble::rownames_to_column()
mypairwise2 %>%
tidyr::pivot_longer(cols = -rowname) %>%
dplyr::filter(complete.cases(.)) %>%
dplyr::mutate(description =
glue::glue(
"The comparison between rowname and name has a p-value of round(value, 2)."
)
) %>%
dplyr::select(description) %>%
dplyr::pull() -> mypairwisedescription
mypairwisedescription <- unlist(mypairwisedescription)
mypairwisedescription <- c(
"In the pairwise comparison of",
mypairwisedescription)📊 Deep Dive: .a.yaml Analysis Definition Architecture
The .a.yaml files define the analysis options and
parameters for jamovi modules. They serve as the configuration blueprint
that controls what options users see and how they interact with your
analysis.
ClinicoPath .a.yaml → self$options Flow
This diagram shows how ClinicoPath .a.yaml definitions map to
accessible self$options values:
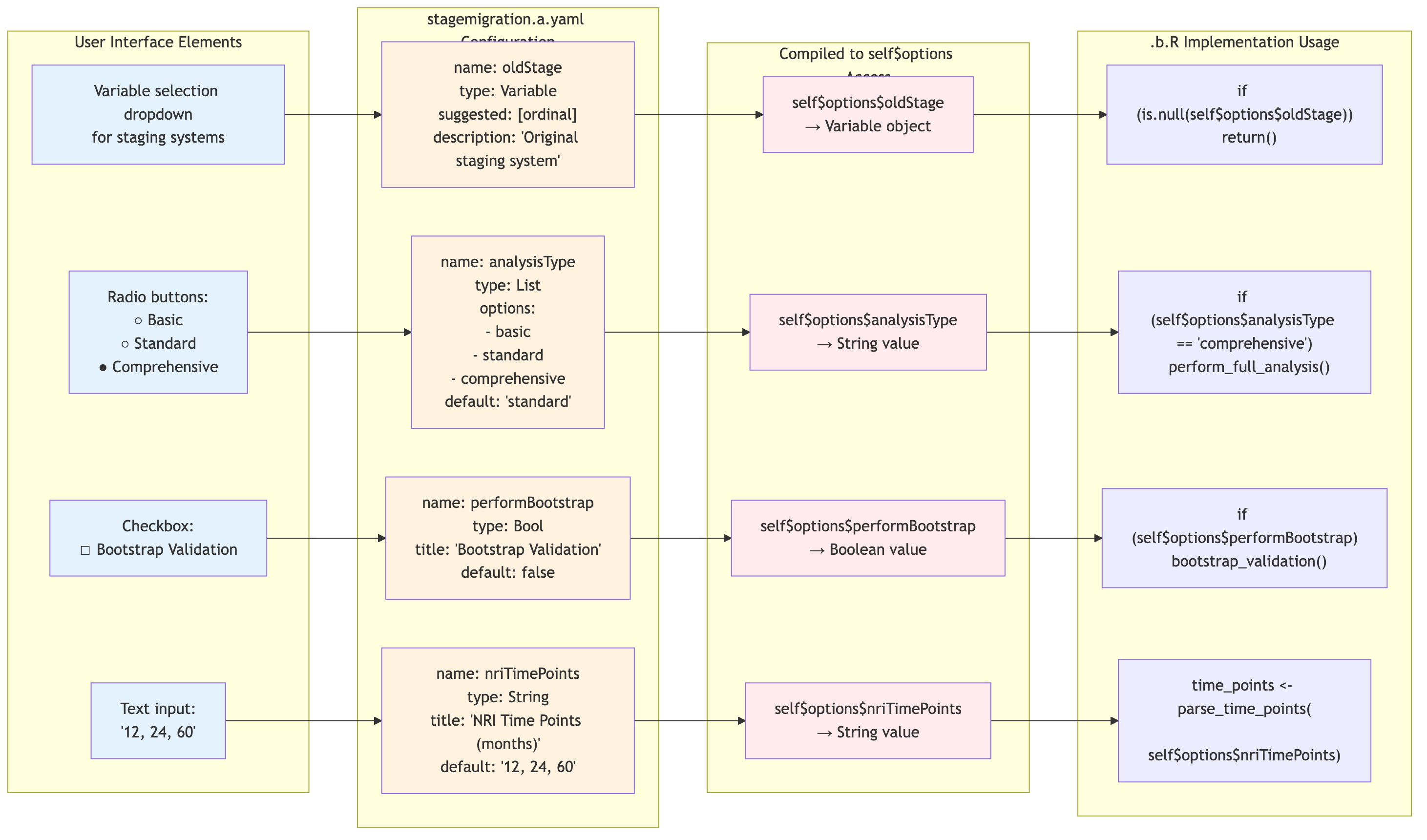
Source:
vignettes/07-a-yaml-options-flow.mmd
ClinicoPath Option Type Decision Tree
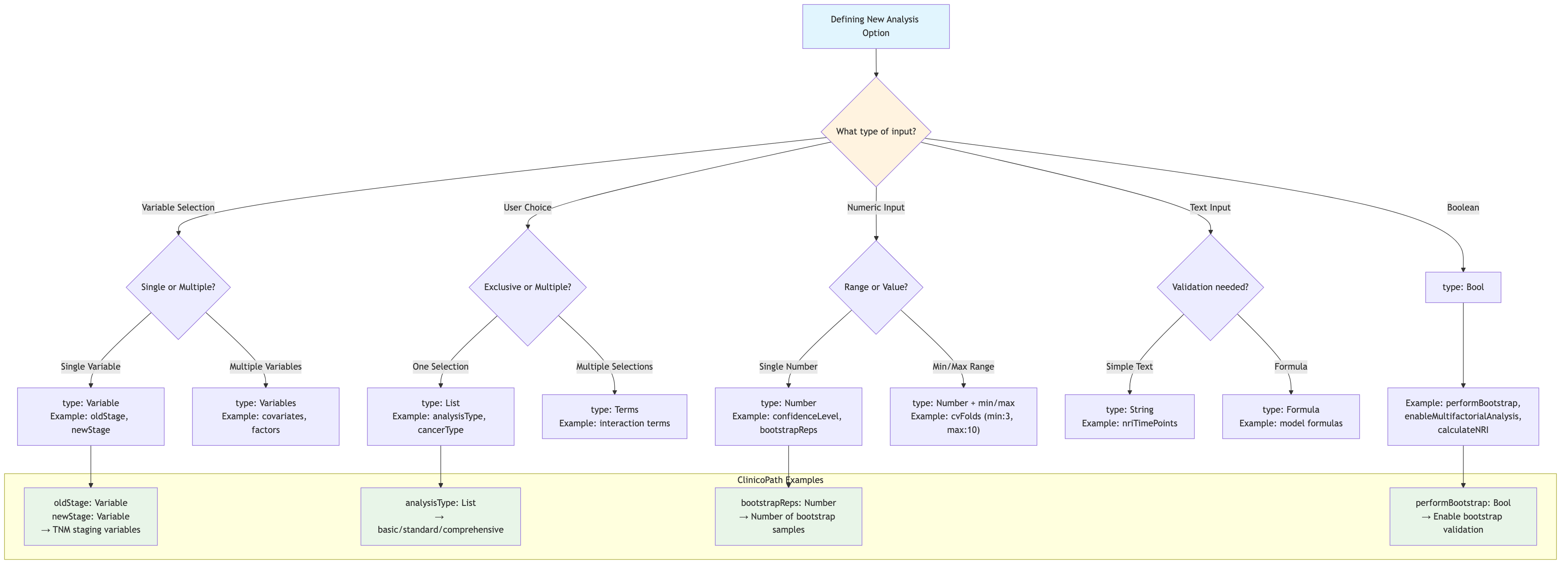
Source:
vignettes/08-option-type-decision-tree.mmd
Complete .a.yaml Anatomy
---
name: jjbarstats # Function identifier (must match .b.R class)
title: Bar Charts # Display name in jamovi menu
menuGroup: JJStatsPlotD # Top-level menu category
menuSubgroup: 'Categorical vs Categorical' # Sub-menu organization
version: '0.0.3' # Module version
jas: '1.2' # jamovi Analysis Specification version
description:
main: | # Main description for help system
'Wrapper Function for ggstatsplot::ggbarstats...'
R: # R-specific documentation
dontrun: true # Skip examples in R CMD check
usage: | # Example usage code
# example will be added
options: # Core configuration section
- name: data # Required data parameter
type: Data
description:
R: >
The data as a data frame.Core Option Types and Patterns
1. Variable Selection Options
Single Variable:
- name: explanatory
title: Explanatory Variable
type: Variable # Single variable selection
suggested: [ ordinal, nominal ] # Preferred variable types
permitted: [ factor ] # Allowed variable types
description:
R: >
The explanatory variable for group comparison.Multiple Variables:
- name: vars
title: Dependent Variable(s)
type: Variables # Multiple variable selection
description: >
The variable(s) that will appear as rows in the cross table.Optional Variables with NULL Default:
2. Level Selection for Categorical Variables
- name: outcomeLevel
title: Event Level
type: Level # Level selector for categorical var
variable: (outcome) # References another option
description:
R: >
The level of the outcome variable that will be used as the event level.
- name: dod
title: Dead of Disease
type: Level
variable: (outcome)
allowNone: true # Allow no selection4. List/Dropdown Options
- name: typestatistics
title: 'Type of Statistic'
type: List
options:
- title: Parametric # User-friendly display name
name: parametric # Internal value
- title: Nonparametric
name: nonparametric
- title: Robust
name: robust
- title: Bayes
name: bayes
default: parametric # Must match one of the 'name' values
- name: analysistype
title: 'Survival Type'
type: List
options:
- title: Overall
name: overall
- title: Cause Specific
name: cause
- title: Competing Risk
name: compete
default: overall5. Numeric Input Options
- name: endplot
title: Plot End Time
type: Integer # Whole numbers only
default: 60
- name: ybegin_plot
title: Start y-axis
type: Number # Decimal numbers allowed
default: 0.00
min: 0.00 # Optional constraints
max: 1.00
- name: rate_multiplier
title: "Rate Multiplier"
type: Integer
default: 100
description: >
Specify the multiplier for incidence rates (e.g., 100 for rates per 100 person-years).Real-World Complex Examples
Example 1: Survival Analysis Configuration
# From survival.a.yaml - Advanced survival analysis options
- name: tint
title: Using Dates to Calculate Survival Time
type: Bool
default: false
description:
R: >
If the time is in date format, select this option to calculate survival time.
- name: dxdate
title: 'Diagnosis Date'
type: Variable
description:
R: >
The date of diagnosis for time calculation.
- name: timetypedata
title: 'Time Type in Data (e.g., YYYY-MM-DD)'
type: List
options:
- title: ymdhms
name: ymdhms
- title: ymd
name: ymd
- title: dmy
name: dmy
default: ymd
description:
R: select the time type in dataExample 2: Statistical Test Configuration
# From jjbarstats.a.yaml - Statistical test options
- name: padjustmethod
title: 'Adjustment Method'
type: List
options:
- title: holm
name: holm
- title: bonferroni
name: bonferroni
- title: BH
name: BH
- title: fdr
name: fdr
- title: none
name: none
default: holm
- name: pairwisedisplay
title: 'Pairwise Display'
type: List
options:
- title: significant
name: significant
- title: non-significant
name: non-significant
- title: everything
name: everything
default: significantBest Practices for .a.yaml Development
1. Variable Type Guidelines
- Use
suggestedto guide users toward appropriate variable types - Use
permittedto enforce data type requirements - Always specify both for Variable and Variables types
2. Default Value Strategy
- Set
default: NULLfor optional parameters - Provide sensible defaults for required parameters
- Use
allowNone: truefor optional Level parameters
Integration with ClinicoPath Module System
1. MenuGroup Organization
menuGroup: SurvivalD # Maps to survival analysis section
menuSubgroup: ClinicoPath Survival
menuSubtitle: 'Univariate Survival Analysis, Cox, Kaplan-Meier'Available MenuGroups in ClinicoPath: - ExplorationD:
Descriptive analysis and data exploration - SurvivalD:
Survival analysis and time-to-event modeling -
JJStatsPlotD: Statistical plots and visualizations -
meddecideT: Medical decision analysis and ROC curves
Common Pitfalls and Solutions
1. Default Value Mismatches
❌ Wrong:
- name: method
type: List
options:
- title: Method A
name: methodA
default: Method A # Should be 'methodA', not 'Method A'✅ Correct:
Testing Your .a.yaml Files
# Always test compilation after .a.yaml changes
jmvtools::prepare()
devtools::document()
# Test in jamovi interface
jmvtools::install()The .a.yaml files are the foundation of user interaction
in jamovi modules. They must be precise, well-documented, and thoroughly
tested to ensure a smooth user experience and proper integration with
the underlying R analysis code.
📋 Deep Dive: .r.yaml Results Definition Architecture
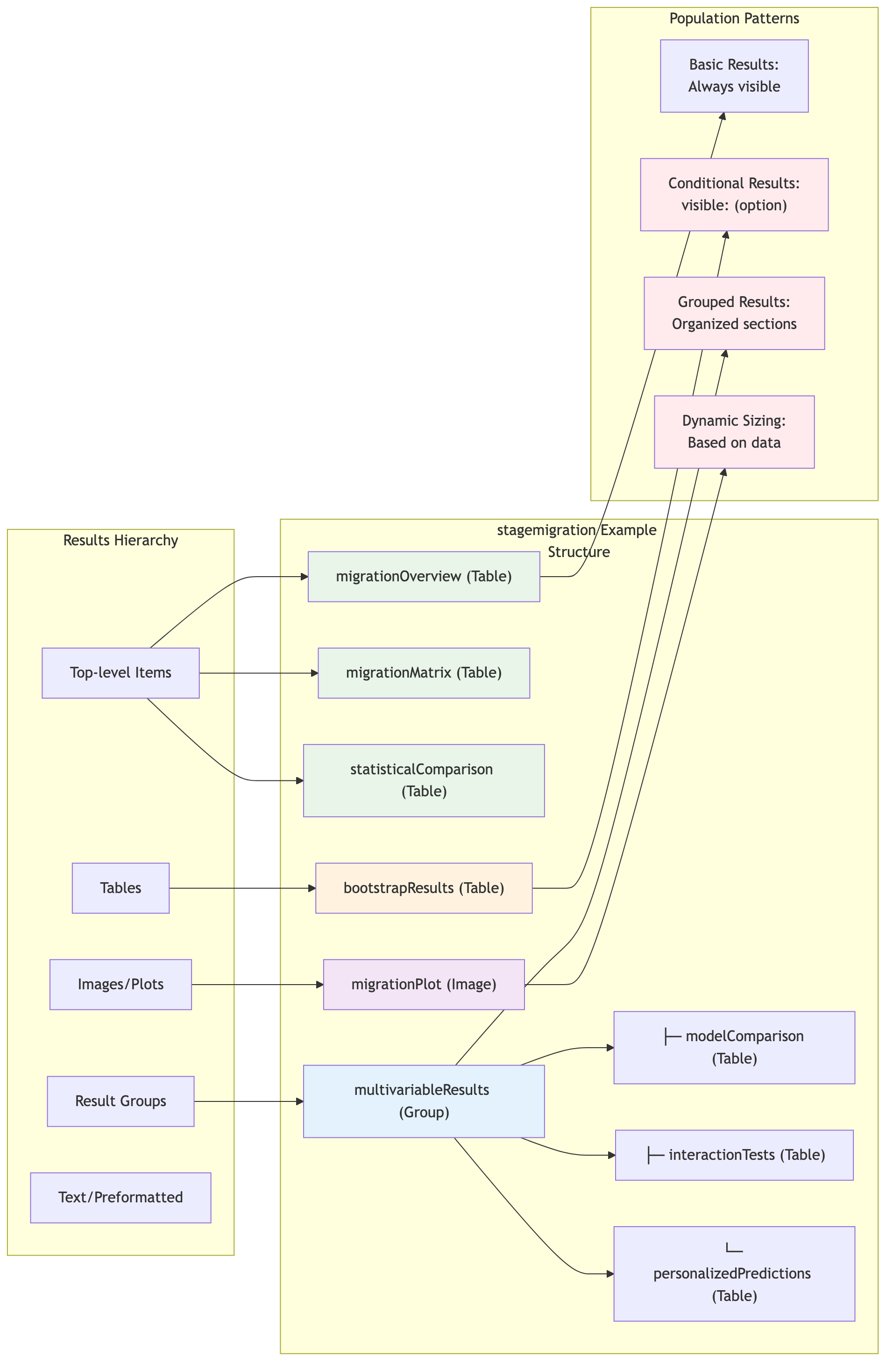
The .r.yaml files define the output structure and
presentation for jamovi modules. They specify what results users will
see, how they’re organized, and how they’re displayed. This is where you
define tables, plots, and text outputs that will be generated by your
analysis.
ClinicoPath self$results → .r.yaml Mapping
This diagram shows how ClinicoPath analyses populate
self$results based on .r.yaml definitions:
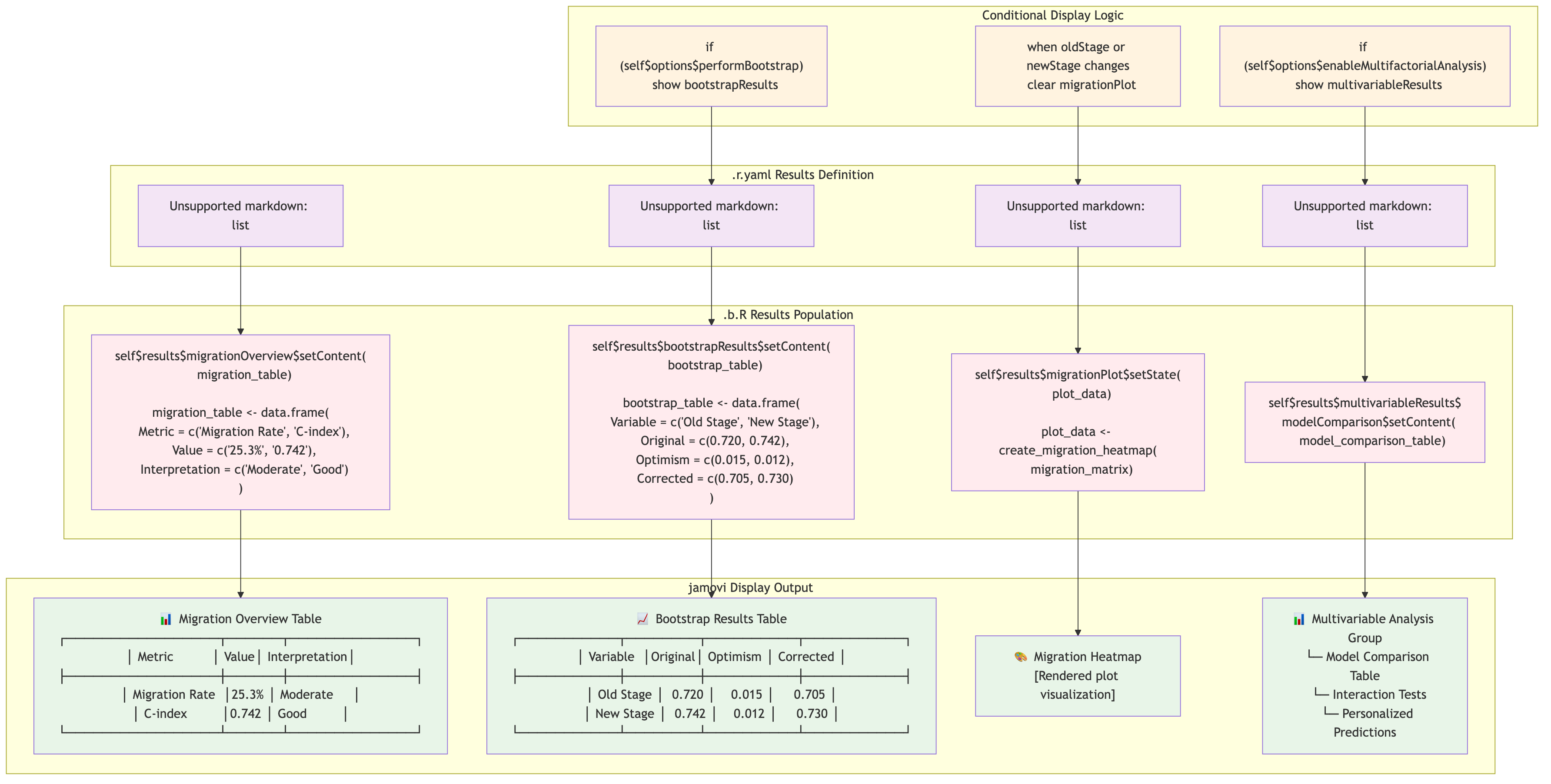
Source:
vignettes/09-results-yaml-mapping.mmd
ClinicoPath Results Organization Pattern
Source:
vignettes/10-results-organization-pattern.mmd
Complete .r.yaml Anatomy
---
name: survival # Must match .a.yaml and .b.R
title: Survival Analysis # Display title for results panel
jrs: '1.1' # jamovi Results Specification version
# Global clear conditions (optional)
clearWith:
- dep
- group
- grvar
items: # Core results structure
- name: subtitle
title: '`Survival Analysis - ${explanatory}`'
type: Preformatted
- name: medianTable
title: '`Median Survival Table: Levels for ${explanatory}`'
type: Table
# ... table definition
refs: # References section
- finalfit
- survival
- survminerCore Result Types and Patterns
1. Text and Preformatted Output
Simple Preformatted Text:
- name: medianSummary
title: '`Median Survival Summary and Table - ${explanatory}`'
type: Preformatted
clearWith:
- explanatory
- outcome
- outcomeLevelHTML Output:
- name: todo
title: To Do
type: Html
clearWith:
- explanatory
- outcome
- name: tablestyle2
title: '`Cross Table - ${group}`'
type: Html
visible: (sty:finalfit) # Conditional visibility
refs: finalfit # Package referenceDynamic Titles with Variable Interpolation:
2. Table Definitions
Basic Table Structure:
- name: medianTable
title: '`Median Survival Table: Levels for ${explanatory}`'
type: Table
rows: 0 # Dynamic row count
columns:
- name: factor
title: "Levels"
type: text
- name: records
title: "Records"
type: integer
- name: median
title: "Median"
type: numberAdvanced Table with Formatting:
- name: survTable
title: '`1, 3, 5 year Survival - ${explanatory}`'
type: Table
rows: 0
columns:
- name: surv
title: "Survival"
type: number
format: pc # Percentage formatting
- name: lower
title: "Lower"
superTitle: '95% Confidence Interval' # Column grouping
type: number
format: pc
- name: upper
title: "Upper"
superTitle: '95% Confidence Interval'
type: number
format: pcTransposed Table:
3. Image/Plot Outputs
Basic Plot Definition:
- name: plot
title: '`Survival Plot - ${explanatory}`'
type: Image
width: 600
height: 450
renderFun: .plot # Function name in .b.R
visible: (sc) # Conditional visibility
requiresData: truePlot with Comprehensive Clear Conditions:
4. Output Variables (Data Addition)
- name: calculatedtime
title: Add Calculated Time to Data
type: Output
varTitle: '`Calculated Time - from ${ dxdate } to { fudate }`'
varDescription: '`Calculated Time from Given Dates`'
measureType: continuous
clearWith:
- tint
- dxdate
- fudate
- name: outcomeredefined
title: Add Redefined Outcome to Data
type: Output
varTitle: '`Redefined Outcome - from ${ outcome }`'
varDescription: Redefined Outcome from Analysis TypeReal-World Complex Examples
Example 1: Survival Analysis Results Structure
# From survival.r.yaml - Comprehensive survival analysis outputs
items:
- name: medianSummary
title: '`Median Survival Summary and Table - ${explanatory}`'
type: Preformatted
clearWith: [explanatory, outcome, outcomeLevel, overalltime]
- name: medianTable
title: '`Median Survival Table: Levels for ${explanatory}`'
type: Table
rows: 0
columns:
- name: factor
title: "Levels"
type: text
- name: records
title: "Records"
type: integer
- name: events
title: "Events"
type: integer
- name: median
title: "Median"
type: number
- name: x0_95lcl
title: "Lower"
superTitle: '95% Confidence Interval'
type: number
- name: x0_95ucl
title: "Upper"
superTitle: '95% Confidence Interval'
type: number
- name: cox_ph
title: 'Proportional Hazards Assumption'
type: Preformatted
visible: (ph_cox) # Only show when option enabled
- name: plot
title: '`Survival Plot - ${explanatory}`'
type: Image
width: 600
height: 450
renderFun: .plot
visible: (sc)
requiresData: trueExample 2: Medical Decision Analysis Results
# From decision.r.yaml - Decision analysis outputs
items:
- name: cTable
title: 'Recoded Data for Decision Test Statistics'
type: Table
rows: 0
columns:
- name: newtest
title: ''
type: text
- name: GP
title: 'Gold Positive'
type: number
- name: GN
title: 'Gold Negative'
type: number
- name: ratioTable
title: ''
type: Table
swapRowsColumns: true
rows: 1
columns:
- name: Sens
title: 'Sensitivity'
type: number
format: pc
- name: Spec
title: 'Specificity'
type: number
format: pc
- name: PPV
title: 'Positive Predictive Value'
type: number
format: pc
- name: LRP
title: Positive Likelihood Ratio
type: number
- name: plot1
title: 'Fagan nomogram'
type: Image
width: 600
height: 450
renderFun: .plot1
requiresData: true
visible: (fagan)
refs: [Fagan, Fagan2]Advanced Patterns and Features
Common Patterns by Analysis Type
Best Practices for .r.yaml Development
1. Result Organization
- Order items logically (instructions → summaries → tables → plots)
- Use descriptive names that reflect the content
- Group related outputs together
2. Clear Conditions Strategy
- Include all relevant options that affect the output
- Be comprehensive but not excessive
- Test thoroughly to ensure proper clearing
Common Pitfalls and Solutions
1. Missing Clear Conditions
❌ Problem: Results don’t update when options change
✅ Solution: Include all relevant options in
clearWith
2. Incorrect Column Types
❌ Problem: Numbers displayed as text ✅
Solution: Use correct type (number,
integer, text)
3. Render Function Mismatch
❌ Problem: Plot doesn’t display ✅
Solution: Ensure renderFun matches
function name in .b.R
The .r.yaml files define the user experience for viewing
results. They must be carefully designed to present information clearly,
update appropriately, and integrate seamlessly with the analysis logic
in the .b.R files.
🎨 Deep Dive: .u.yaml User Interface Definition Architecture
The .u.yaml files define the user interface layout and
interaction patterns for jamovi modules. They specify how options are
organized, displayed, and interact with each other in the analysis
panel. This is where you create the user experience that researchers
will interact with.
Complete .u.yaml Anatomy
title: Survival Analysis # Display title in jamovi menu
name: survival # Must match .a.yaml, .b.R, .r.yaml
jus: '3.0' # jamovi UI Specification version
stage: 0 # Development stage (0=development, 1=release)
compilerMode: tame # Compiler optimization mode
children: # Root UI container
- type: VariableSupplier # Main variable selection area
persistentItems: false # Don't persist selections across datasets
stretchFactor: 1 # Layout weight
children:
- type: TargetLayoutBox # Variable drop zone
label: Time Elapsed
children:
- type: VariablesListBox
name: elapsedtime # Must match .a.yaml option name
maxItemCount: 1 # Single variable only
isTarget: true # Can receive dropped variablesCore UI Component Types and Patterns
1. Variable Selection Components
Variable Supplier (Main Container):
- type: VariableSupplier
persistentItems: false # Reset on new dataset
stretchFactor: 1 # Layout proportion
children:
- type: TargetLayoutBox # Variable category container
label: Dependent Variable
children:
- type: VariablesListBox # Actual variable selector
name: vars # Links to .a.yaml option
isTarget: true # Accepts dropped variables
maxItemCount: 1 # Single variable limitMultiple Variables vs Single Variable:
# Multiple variables allowed
- type: VariablesListBox
name: dep
isTarget: true # No maxItemCount = unlimited
# Single variable only
- type: VariablesListBox
name: group
maxItemCount: 1 # Restricts to one variable
isTarget: trueLevel Selectors for Categorical Variables:
2. Organizational Components
Collapsible Sections:
- type: CollapseBox
label: Advanced Elapsed Time Options
collapsed: true # Starts collapsed
stretchFactor: 1 # Layout weight
children:
# ... nested componentsLayout Containers:
- type: LayoutBox
margin: large # Spacing around contents
fitToGrid: true # Align to grid
stretchFactor: 1 # Layout weight
children:
# ... child componentsLabels and Groups:
3. Input Controls
Checkboxes:
- type: CheckBox
name: tint # Links to .a.yaml option
fitToGrid: true
# Checkbox with nested controls
- type: CheckBox
name: pairwisecomparisons
children:
- type: ComboBox
name: pairwisedisplay
enable: (pairwisecomparisons) # Only enabled when parent checkedDropdown Menus:
Text Input Boxes:
- type: TextBox
name: landmark
format: number # Numeric input only
enable: (uselandmark) # Conditional enabling
- type: TextBox
name: cutp
format: string # Text input
width: large # Wider input boxOutput Variables:
Real-World Complex Examples
Example 1: Survival Analysis UI Structure
# From survival.u.yaml - Comprehensive survival analysis interface
children:
# Main variable selection area
- type: VariableSupplier
persistentItems: false
stretchFactor: 1
children:
- type: TargetLayoutBox
label: Time Elapsed
children:
- type: VariablesListBox
name: elapsedtime
maxItemCount: 1
isTarget: true
- type: TargetLayoutBox
label: Outcome
children:
- type: VariablesListBox
name: outcome
maxItemCount: 1
isTarget: true
fitToGrid: true
- type: LevelSelector
name: outcomeLevel
enable: (outcome && !multievent)
# Advanced options in collapsible sections
- type: CollapseBox
label: Advanced Elapsed Time Options
collapsed: true
children:
- type: CheckBox
name: tint
- type: ComboBox
name: timetypedata
enable: (tint)
- type: CollapseBox
label: Analysis with Multiple Outcomes
collapsed: true
children:
- type: CheckBox
name: multievent
enable: (outcome)
- type: LevelSelector
name: dod
enable: (outcome && multievent)Example 2: Statistical Test Configuration
# From jjbarstats.u.yaml - Bar chart analysis interface
children:
- type: VariableSupplier
children:
- type: TargetLayoutBox
label: Dependent Variable
children:
- type: VariablesListBox
name: dep
isTarget: true
- type: TargetLayoutBox
label: Split By (Optional)
children:
- type: VariablesListBox
name: grvar
maxItemCount: 1
isTarget: true
- type: CollapseBox
label: Analysis
collapsed: false # Starts expanded
children:
- type: ComboBox
name: typestatistics
- type: CheckBox
name: pairwisecomparisons
children: # Nested controls
- type: ComboBox
name: pairwisedisplay
enable: (pairwisecomparisons)
- type: ComboBox
name: padjustmethod
enable: (pairwisecomparisons)Example 3: Medical Decision Analysis
# From decision.u.yaml - Decision analysis interface
children:
- type: VariableSupplier
children:
- type: TargetLayoutBox
label: Golden Standard
children:
- type: VariablesListBox
name: gold
maxItemCount: 1
isTarget: true
- type: LevelSelector
name: goldPositive
enable: (gold) # Only enabled when variable selected
- type: TargetLayoutBox
label: New Test
children:
- type: VariablesListBox
name: newtest
maxItemCount: 1
isTarget: true
- type: LevelSelector
name: testPositive
enable: (newtest)
- type: Label
label: Prior Probability
children:
- type: CheckBox
name: pp
enable: (!ci) # Disabled when ci is checked
- type: TextBox
name: pprob
format: number
enable: (!ci && pp) # Multiple conditionsAdvanced UI Patterns and Features
1. Conditional Enabling
# Simple condition
enable: (outcome) # Enabled when outcome is selected
# Multiple conditions with AND
enable: (outcome && multievent) # Both conditions must be true
# Multiple conditions with OR
enable: (sty:finalfit || sty:nejm) # Either condition can be true
# Negation
enable: (!ci) # Enabled when ci is NOT checked
# Complex conditions
enable: (!ci && pp) # NOT ci AND pp is checkedUI Organization Patterns by Analysis Type
2. Complex Multi-Step Analysis
children:
- type: VariableSupplier # Main variables
- type: CollapseBox # Advanced data options
label: Advanced Options
collapsed: true
- type: CollapseBox # Multiple outcomes
label: Multiple Outcomes
collapsed: true
- type: CollapseBox # Pairwise comparisons
label: Pairwise Comparisons
collapsed: true
- type: CollapseBox # Plots
label: Plots
collapsed: trueIntegration with ClinicoPath Module System
1. Standard Layout Patterns
- Variable Supplier: Always first, contains main variable selections
- Collapsible Sections: Group related options, start collapsed for advanced features
- Logical Grouping: Data → Analysis → Visualization → Advanced
Best Practices for .u.yaml Development
1. Layout Organization
- Place most important options at the top
- Group related options together
- Use descriptive labels
- Start advanced sections collapsed
Common Pitfalls and Solutions
1. Name Mismatches
❌ Problem: UI component doesn’t link to option ✅
Solution: Ensure name exactly matches
.a.yaml option name
2. Incorrect Conditional Logic
❌ Problem: Components enabled/disabled incorrectly ✅ Solution: Test all condition combinations thoroughly
3. Poor Layout Organization
❌ Problem: Confusing or cluttered interface ✅ Solution: Use logical grouping and collapsible sections
4. Missing Variable Constraints
❌ Problem: Users can select inappropriate variable
types ✅ Solution: Use maxItemCount and
proper suggested/permitted in .a.yaml
The .u.yaml files define the user interface that
researchers interact with. They must be intuitive, responsive, and guide
users toward making appropriate analysis choices while preventing
invalid option combinations.
Diagrams
eval=FALSE, include=FALSE, echo=FALSE
DiagrammeR::mermaid(
diagram = here::here("vignettes/graph.mmd"),
height = 200
)
Remotes:
easystats/correlation,
easystats/report
# Future Works:
## ndphillips/FFTrees
# gtsummary
# myvars <- jmvcore::constructFormula(terms = self$options$vars)
# myvars <- jmvcore::decomposeFormula(formula = myvars)
# myvars <- unlist(myvars)
# mytableone2 <- self$data %>%
# dplyr::select(myvars)
# mytableone2 <- gtsummary::tbl_summary(mytableone2)
# self$results$text2$setContent(mytableone2)
# - name: outcomeLevel
# title: |
# Select Event (Death, Recurrence)
# type: Level
# variable: (outcome)
,
arsenal,
rlang,
knitr,
remotes,
kableExtra,
caret,
irr
Remotes:
easystats/bayestestR,
easystats/performance,
easystats/parameters,
easystats/report
Suggests:
effectsize,
emmeans,
rmarkdown,
igraph,
iterators,
rms,
commonmark,
sass
# #
# #
# # if (is.null(self$options$dep) || is.null(self$options$group))
# # return()
# #
# # mydata <- self$data
# #
# # mydep <- self$data[[self$options$dep]]
# #
# # mygroup <- self$data[[self$options$group]]
# #
# #
# # # klass <- print(
# # # list(
# # # "mydep" = c(typeof(mydep), class(mydep)),
# # # "mygroup" = c(typeof(mygroup), class(mygroup))
# # # )
# # # )
# #
# #
# # # self$results$text1$setContent(klass)
# #
# #
# # # plotData <- data.frame(gr = mygroup,
# # # dp = mydep)
# # # plotData <- jmvcore::naOmit(plotData)
# # # mydata_changes <- plotData %>%
# # # dplyr::group_by(gr, dp) %>%
# # # dplyr::tally(x = .)
# # #
# # # self$results$text2$setContent(mydata_changes)
# # #
# # # plotData <- data.frame(gr = mygroup,
# # # dp = mydep)
# # #
# # # plotData <- jmvcore::naOmit(plotData)
# # #
# # #
# # # mydata_changes <- plotData %>%
# # # dplyr::group_by(gr, dp) %>%
# # # dplyr::tally(x = .)
# # #
# # #
# # # deneme <- ggalluvial::is_alluvia_form(
# # # as.data.frame(mydata_changes),
# # # axes = 1:2, silent = TRUE)
# #
# # # nodes = data.frame("name" =
# # # c(self$options$group,
# # # self$options$dep))
# # #
# # # links <- mydata_changes
# # #
# # # names(links) = c("source", "target", "value")
# # #
# # # deneme <- networkD3::sankeyNetwork(Links = links, Nodes = nodes,
# # # Source = "source", Target = "target",
# # # Value = "value", NodeID = "name",
# # # fontSize= 12, nodeWidth = 30)
# #
# #
# #
# # # self$results$text3$setContent(deneme)
# #
# #
# #
# #
# # # Prepare Data for Plot ----
# #
# # direction <- self$options$direction
# #
# # mydata <- self$data
# #
# # mydep <- self$data[[self$options$dep]]
# #
# # mygroup <- self$data[[self$options$group]]
# #
# # contin <- c("integer", "numeric", "double")
# # categ <- c("factor")
# #
# # # independent, factor, continuous ----
# # # ggbetweenstats violin plots for comparisons between groups/conditions
# # if (direction == "independent" && class(mygroup) == "factor" && class(mydep) %in% contin) {
# # plotData <- data.frame(gr = mygroup,
# # dp = jmvcore::toNumeric(mydep))
# #
# #
# #
# #
# # # independent, continuous, continuous ----
# # # ggscatterstats scatterplots for correlations between two variables
# #
# # if (direction == "independent" && class(mygroup) %in% contin && class(mydep) %in% contin) {
# # plotData <- data.frame(gr = jmvcore::toNumeric(mygroup),
# # dp = jmvcore::toNumeric(mydep))
# #
# #
# #
# #
# #
# # # independent, factor, factor ----
# # # ggbarstats bar charts for categorical data
# # if (direction == "independent" && class(mygroup) == "factor" && class(mydep) == "factor") {
# #
# # plotData <- data.frame(gr = mygroup,
# # dp = mydep)
# #
# #
# #
# # # independent, continuous, factor ----
# #
# # if (direction == "independent" && class(mygroup) %in% contin && class(mydep) == "factor") {
# #
# # stop("Please switch the values: factor variable should be on x-axis and continuous variable should be on y-axis")
# # }
# #
# #
# #
# # # repeated, factor, continuous ----
# # # ggwithinstats violin plots for comparisons within groups/conditions
# #
# #
# #
# # if (direction == "repeated" && class(mygroup) == "factor" && class(mydep) %in% contin) {
# # plotData <- data.frame(gr = mygroup,
# # dp = jmvcore::toNumeric(mydep))
# #
# #
# #
# #
# # # repeated, continuous, continuous ----
# # # rmcorr::rmcorr()
# #
# #
# # if (direction == "repeated" && class(mygroup) %in% contin && class(mydep) %in% contin) {
# #
# #
# # stop("Currently this module does not support repeated measures correlation.")
# #
# # }
# #
# #
# # # repeated, factor, factor ----
# # # http://corybrunson.github.io/ggalluvial/
# #
# # if (direction == "repeated" && class(mygroup) == "factor" && class(mydep) == "factor") {
# # plotData <- data.frame(gr = mygroup,
# # dp = mydep)
# #
# #
# #
# # # repeated, continuous, factor ----
# #
# # if (direction == "repeated" && class(mygroup) %in% contin && class(mydep) == "factor") {
# #
#
#
#
#
#
# # Results ----
#
#
#
# # Send Data to Plot ----
#
# # plotData <- jmvcore::naOmit(plotData)
# # image <- self$results$plot
# # image$setState(plotData)
#
#
# # }
#
#
# # ,
# #
# # .plot = function(image, ...) { # <-- the plot function ----
# #
# #
# # if (is.null(self$options$dep) || is.null(self$options$group))
# # return()
# #
# #
# # plotData <- image$state
# #
# # direction <- self$options$direction
# #
# # mydata <- self$data
# #
# # mydep <- self$data[[self$options$dep]]
# #
# # mygroup <- self$data[[self$options$group]]
# #
# # contin <- c("integer", "numeric", "double")
# # categ <- c("factor")
# #
# # # independent, factor, continuous ----
# # # ggbetweenstats violin plots for comparisons between groups/conditions
# #
# # if (direction == "independent" && class(mygroup) == "factor" && class(mydep) %in% contin) {
# #
# # plot <- ggstatsplot::ggbetweenstats(
# # data = plotData,
# # x = gr,
# # y = dp
# # )
# # }
# #
# # # independent, continuous, continuous ----
# # # ggscatterstats scatterplots for correlations between two variables
# #
# #
# # if (direction == "independent" && class(mygroup) %in% contin && class(mydep) %in% contin) {
# #
# # plot <- ggstatsplot::ggscatterstats(
# # data = plotData,
# # x = gr,
# # y = dp
# # )
# #
# # }
# #
# # # independent, factor, factor ----
# # # ggbarstats bar charts for categorical data
# #
# #
# # if (direction == "independent" && class(mygroup) == "factor" && class(mydep) == "factor") {
# #
# #
# #
# # plot <- ggstatsplot::ggbarstats(
# # data = plotData,
# # main = gr,
# # condition = dp
# # )
# # }
# #
# # # repeated, factor, continuous ----
# # # ggwithinstats violin plots for comparisons within groups/conditions
# #
# #
# # if (direction == "repeated" && class(mygroup) == "factor" && class(mydep) %in% contin) {
# #
# #
# # plot <- ggstatsplot::ggwithinstats(
# # data = plotData,
# # x = gr,
# # y = dp
# # )
# #
# # }
# #
# # # repeated, continuous, continuous ----
# # # rmcorr::rmcorr()
# #
# # # my.rmc <- rmcorr::rmcorr(participant = Subject,
# # # measure1 = PacO2,
# # # measure2 = pH,
# # # dataset = rmcorr::bland1995)
# # #
# # # plot(my.rmc, overall = TRUE)
# # #
# # # ggplot2::ggplot(rmcorr::bland1995,
# # # ggplot2::aes(x = PacO2,
# # # y = pH,
# # # group = factor(Subject),
# # # color = factor(Subject)
# # # )
# # # ) +
# # # ggplot2::geom_point(ggplot2::aes(colour = factor(Subject))) +
# # # ggplot2::geom_line(ggplot2::aes(y = my.rmc$model$fitted.values), linetype = 1)
# #
# #
# #
# # # repeated, factor, factor ----
# # # http://corybrunson.github.io/ggalluvial/
# # # networkD3
# #
# #
# # if (direction == "repeated" && class(mygroup) == "factor" && class(mydep) == "factor") {
# #
# #
# # mydata_changes <- plotData %>%
# # dplyr::group_by(gr, dp) %>%
# # dplyr::tally(x = .)
# #
# #
# # # head(as.data.frame(UCBAdmissions), n = 12)
# #
# # # ggalluvial::is_alluvia_form(
# # # as.data.frame(UCBAdmissions),
# # # axes = 1:3, silent = TRUE)
# #
# #
# #
# # # plot <- ggplot(as.data.frame(UCBAdmissions),
# # # aes(y = Freq, axis1 = Gender, axis2 = Dept)) +
# # # geom_alluvium(aes(fill = Admit), width = 1/12) +
# # # geom_stratum(width = 1/12, fill = "black", color = "grey") +
# # # geom_label(stat = "stratum", infer.label = TRUE) +
# # # scale_x_discrete(limits = c("Gender", "Dept"), expand = c(.05, .05)) +
# # # scale_fill_brewer(type = "qual", palette = "Set1") +
# # # ggtitle("UC Berkeley admissions and rejections, by sex and department")
# #
# #
# #
# #
# #
# # stratum <- ggalluvial::StatStratum
# #
# # plot <- ggplot2::ggplot(data = mydata_changes,
# # ggplot2::aes(axis1 = gr,
# # axis2 = dp,
# # y = n)) +
# # ggplot2::scale_x_discrete(limits = c(self$options$group, self$options$dep),
# # expand = c(.1, .05)
# # ) +
# # ggplot2::xlab(self$options$group) +
# # ggalluvial::geom_alluvium(ggplot2::aes(fill = gr,
# # colour = gr
# # )) +
# # ggalluvial::geom_stratum() +
# # ggalluvial::stat_stratum(geom = "stratum") +
# # ggplot2::geom_label(stat = stratum, infer.label = TRUE) +
# #
# # # ggalluvial::geom_stratum(stat = "stratum", label.strata = TRUE) +
# # # ggplot2::geom_text(stat = "stratum", infer.label = TRUE) +
# # # ggplot2::geom_text(label.strata = TRUE) +
# # # ggalluvial::geom_stratum()
# # ggplot2::theme_minimal()
# # # ggplot2::ggtitle(paste0("Changes in ", self$options$group))
# # #
# # #
# # # nodes = data.frame("name" =
# # # c(self$options$group,
# # # self$options$dep))
# # #
# # # links <- mydata_changes
# # #
# # # names(links) = c("source", "target", "value")
# # #
# # # plot <- networkD3::sankeyNetwork(Links = links, Nodes = nodes,
# # # Source = "source", Target = "target",
# # # Value = "value", NodeID = "name",
# # # fontSize= 12, nodeWidth = 30)
# #
# # # library(networkD3)
# # # nodes = data.frame("name" =
# # # c("Node A", # Node 0
# # # "Node B", # Node 1
# # # "Node C", # Node 2
# # # "Node D"))# Node 3
# # # links = as.data.frame(matrix(c(
# # # 0, 1, 10, # Each row represents a link. The first number
# # # 0, 2, 20, # represents the node being conntected from.
# # # 1, 3, 30, # the second number represents the node connected to.
# # # 2, 3, 40),# The third number is the value of the node
# # # byrow = TRUE, ncol = 3))
# # # names(links) = c("source", "target", "value")
# # # sankeyNetwork(Links = links, Nodes = nodes,
# # # Source = "source", Target = "target",
# # # Value = "value", NodeID = "name",
# # # fontSize= 12, nodeWidth = 30)
# #
# # # plot <- c("Under Construction")
# #
# # # plot <- list(plot1,
# # # plot2)
# #
# #
# #
# # }
# #
# #
# #
# # print(plot)
# # TRUE
# #
# # }
# #
# # )
# # )
# Packages
Imports:
jmvcore (>= 0.8.5),
R6,
dplyr,
survival,
survminer,
finalfit,
arsenal,
purrr,
glue,
janitor,
ggplot2,
forcats,
ggstatsplot,
tableone,
explore,
tangram,
irr,
rlang,
tidyselect,
knitr
Remotes:
easystats/correlation,
neuropsychology/psycho.R@0.4.0
Suggests:
rmarkdown,
remotes,
devtools,
lubridate,
broom,
GGally,
gridExtra,
Hmisc,
lme4,
magrittr,
mice,
pillar,
pROC,
scales,
stringr,
tibble,
tidyr,
covr,
cmprsk,
readr,
rstan,
survey,
testthat,
backports,
generics,
assertthat,
pkgconfig,
Rcpp,
BH,
plogr,
ellipsis,
gtable,
progress,
RColorBrewer,
reshape,
digest,
lazyeval,
viridisLite,
withr,
Formula,
latticeExtra,
acepack,
data.table,
htmlTable,
viridis,
htmltools,
base64enc,
minqa,
nloptr,
RcppEigen,
mitml,
cli,
crayon,
fansi,
utf8,
vctrs,
farver,
labeling,
munsell,
lifecycle,
stringi,
ggpubr,
maxstat,
survMisc,
jsonlite,
rex,
evaluate,
highr,
markdown,
xfun,
hms,
clipr,
mime,
tinytex,
StanHeaders,
inline,
loo,
pkgbuild,
numDeriv,
mitools,
pkgload,
praise,
zeallot,
colorspace,
prettyunits,
checkmate,
htmlwidgets,
pan,
jomo,
ordinal,
ucminf,
ggrepel,
ggsci,
cowplot,
ggsignif,
polynom,
exactRankTests,
mvtnorm,
KMsurv,
zoo,
km.ci,
xtable,
curl,
openssl,
askpass,
sys,
matrixStats,
callr,
desc,
rprojroot,
processx,
ps,
DBI,
png,
jpeg,
boot,
grid,
snakecase,
caret,
iterators,
timeDate,
foreach,
plyr,
ModelMetrics,
nlme,
reshape2,
recipes,
BradleyTerry2,
e1071,
earth,
fastICA,
gam,
ipred,
kernlab,
klaR,
MASS,
ellipse,
mda,
mgcv,
mlbench,
MLmetrics,
nnet,
party,
pls,
proxy,
randomForest,
RANN,
spls,
subselect,
pamr,
superpc,
Cubist,
rpart,
qgraph,
nFactors,
ppcor,
rstanarm,
MuMIn,
blavaan,
# Develop
# install.packages('jmvtools', repos=c('https://repo.jamovi.org', 'https://cran.r-project.org'))
# jmvtools::check("C://Program Files//jamovi//bin")
# jmvtools::install(home = "C://Program Files//jamovi//bin")
#
# jmvtools::install(pkg = "C://ClinicoPath", home = "C://Program Files//jamovi//bin")
# devtools::build(path = "C:\\ClinicoPathOutput")
# .libPaths(new = "C:\\ClinicoPathLibrary")
# devtools::build(path = "C:\\ClinicoPathOutput", binary = TRUE, args = c('--preclean'))
Sys.setenv(TZ = "Europe/Istanbul")
library("jmvtools")
jmvtools::check()
# rhub::check_on_macos()
# rhub::check_for_cran()
# codemetar::write_codemeta()
devtools::check()
# From CRAN
# install.packages("attachment")
# From github
# remotes::install_github("ThinkR-open/attachment")
# If you correctly called the package dependencies in the {roxygen2} skeleton, in your functions, in your Rmarkdown vignettes and in your tests, you only need to run attachment::att_to_description()just before devtools::check(). And that’s it, there is nothing else to remember !
# attachment::att_to_description()
devtools::document()
codemetar::write_codemeta()
# rmarkdown::render('/Users/serdarbalciold/histopathRprojects/ClinicoPath/README.Rmd', encoding = 'UTF-8', knit_root_dir = '~/histopathRprojects/ClinicoPath', quiet = TRUE)
pkgdown::build_articles()
# pkgdown::build_favicons()
pkgdown::build_home()
pkgdown::build_news()
pkgdown::build_reference()
# pkgdown::build_reference_index()
# pkgdown::build_tutorials()
pkgdown::build_site()
# devtools::github_release()
# gitUpdateCommitPush
CommitMessage <- paste("updated on ", Sys.time(), sep = "")
wd <- getwd()
gitCommand <- paste("cd ", wd, " \n git add . \n git commit --message '", CommitMessage, "' --no-verify \n git push origin master \n", sep = "")
# gitCommand <- paste("cd ", wd, " \n git add . \n git commit --no-verify --message '", CommitMessage, "' \n git push origin master \n", sep = "")
system(command = gitCommand, intern = TRUE)
# jmvtools::install()
#
# jmvtools::create('ClinicoPath') # Use ClinicoPath instead of SuperAwesome
#
# jmvtools::addAnalysis(name='ttest', title='Independent Samples T-Test')
#
# jmvtools::addAnalysis(name='survival', title='survival')
#
# jmvtools::addAnalysis(name='correlation', title='correlation')
#
# jmvtools::addAnalysis(name='tableone', title='TableOne')
#
# jmvtools::addAnalysis(name='crosstable', title='CrossTable')
#
#
# jmvtools::addAnalysis(name='writesummary', title='WriteSummary')
# jmvtools::addAnalysis(name='finalfit', title='FinalFit')
# jmvtools::addAnalysis(name='multisurvival', title='FinalFit Multivariate Survival')
# jmvtools::addAnalysis(name='report', title='Report General Features')
# jmvtools::addAnalysis(name='frequencies', title='Frequencies')
# jmvtools::addAnalysis(name='statsplot', title='GGStatsPlot')
# jmvtools::addAnalysis(name='statsplot2', title='GGStatsPlot2')
# jmvtools::addAnalysis(name='statsplotbetween', title='Stats Plot Between')
# jmvtools::addAnalysis(name='competingsurvival', title='Competing Survival')
# jmvtools::addAnalysis(name='scat2', title='scat2')
# jmvtools::addAnalysis(name='decisioncalculator', title='Decision Calculator')
# jmvtools::addAnalysis(name='agreement', title='Interrater Intrarater Reliability')
# jmvtools::addAnalysis(name='cluster', title='Cluster Analysis')
# jmvtools::addAnalysis(name='tree', title='Decision Tree')
#
# jmvtools::addAnalysis(name='oddsratio', title='Odds Ratio Table and Plot')
# jmvtools::addAnalysis(name='roc', title='ROC')
# jmvtools::addAnalysis(name = "icccoeff", title = "ICC coefficients")
# jmvtools::addAnalysis(name = "gtsummary", title = "Tables via gtsummary")
# jmvtools::addAnalysis(name = "alluvial", title = "Alluvial Diagrams")
Sys.unsetenv("R_PROFILE_USER")
devtools::check()
devtools::install()
# jmvtools::check()
jmvtools::install()
formula <- jmvcore::constructFormula(terms = c("A", "B", "C"), dep = "D")
jmvcore::constructFormula(terms = list("A", "B", c("C", "D")), dep = "E")
jmvcore::constructFormula(terms = "A B")
jmvcore::constructFormula(terms = list("A", "B", "C"))
vars <- jmvcore::decomposeFormula(formula = formula)
unlist(vars)
cformula <- jmvcore::composeTerm(components = formula)
jmvcore::composeTerm("A B")
jmvcore::composeTerm(components = c("A", "B", "C"))
jmvcore::decomposeTerm(term = c("A", "B", "C"))
jmvcore::decomposeTerm(term = formula)
jmvcore::decomposeTerm(term = cformula)
composeTerm <- jmvcore::composeTerm(components = c("A", "B", "C"))
jmvcore::decomposeTerm(term = composeTerm)
BreastCancer <- readr::read_csv(file = "/Users/serdarbalciold/histopathRprojects/ClinicoPath/data/BreastCancer.csv")
usethis::use_data(BreastCancer)
BreastCancer <- readr::read_csv(file = "/Users/serdarbalciold/histopathRprojects/ClinicoPath/data/BreastCancer.csv")
usethis::use_data(BreastCancer)
colon <- readr::read_csv(file =
"/Users/serdarbalciold/histopathRprojects/ClinicoPath/data/colon.csv")
usethis::use_data(colon)
melanoma <- readr::read_csv(file =
"/Users/serdarbalciold/histopathRprojects/ClinicoPath/data/melanoma.csv")
usethis::use_data(melanoma)
rocdata <- readr::read_csv(file =
"/Users/serdarbalciold/histopathRprojects/ClinicoPath/data/rocdata.csv")
usethis::use_data(rocdata)
histopathology <- readr::read_csv(file =
"/Users/serdarbalciold/histopathRprojects/ClinicoPath/data/histopathology.csv")
usethis::use_data(histopathology)
## force git
# gitUpdateCommitPush
CommitMessage <- paste("updated on ", Sys.time(), sep = "")
wd <- getwd()
gitCommand <- paste("cd ", wd, " \n git add . \n git commit --message '", CommitMessage, "' \n git push origin master \n", sep = "")
system(command = gitCommand, intern = TRUE)
## update project for release
readyfunctions <- c(
"refs",
# "^agreement",
# "^competingsurvival",
# "^correlation",
"^crosstable",
# "^decision",
# "^decisioncalculator",
# "^icccoeff",
"^multisurvival",
"^oddsratio",
# "^pairchi2",
"^reportcat",
# "^roc",
"^statsplot2",
"^summarydata",
"^survival",
"^tableone"
# "^tree",
# "^utils-pipe"
# "^vartree"
)
readyfunctions <- paste0(readyfunctions, collapse = "|")
files_R <-
list.files(path = here::here("R"),
pattern = readyfunctions,
full.names = TRUE)
files_jamovi <-
list.files(
path = here::here("jamovi"),
pattern = readyfunctions,
full.names = TRUE
)
files_data <-
list.files(
path = here::here("data"),
full.names = TRUE
)
file.copy(from = files_R,
to = "~/ClinicoPath/R/",
overwrite = TRUE)
file.copy(from = files_jamovi,
to = "~/ClinicoPath/jamovi/",
overwrite = TRUE)
file.copy(from = files_data,
to = "~/ClinicoPath/data/",
overwrite = TRUE)
file.copy(from = files_data,
to = "~/histopathRprojects/ClinicoPath/inst/extdata/",
overwrite = TRUE)
# Example
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
library(magrittr)
corx <- deneme %>%
dplyr::select(Age, OverallTime) %>%
stats::cor(method = "spearman") %>%
report::report()
inherits(deneme$Sex, "character")
ggstatsplot::ggbetweenstats(data = deneme,
x = Sex,
y = Age,
type = "p")
ClinicoPath::statsplot2(
data = deneme,
dep = Age,
group = Sex)
devtools::install(upgrade = FALSE, quick = TRUE)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
# library("ClinicoPath")
deneme$Age <- as.numeric(as.character(deneme$Age))
ClinicoPath::writesummary(data = deneme, vars = Age)
ggstatsplot::normality_message(deneme$Age, "Age")
ClinicoPath::writesummary(
data = deneme,
vars = Age)
devtools::install(upgrade = FALSE, quick = TRUE)
library(dplyr)
library(survival)
library(finalfit)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
ClinicoPath::finalfit(data = deneme,
explanatory = Sex,
outcome = Outcome,
overalltime = OverallTime)
devtools::install(upgrade = FALSE, quick = TRUE)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
ClinicoPath::decision(
data = deneme,
gold = Outcome,
goldPositive = "1",
newtest = Smoker,
testPositive = "TRUE")
ClinicoPath::decision(
data = deneme,
gold = LVI,
goldPositive = "Present",
newtest = PNI,
testPositive = "Present")
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
ggstatsplot::ggbetweenstats(data = deneme,
x = LVI,
y = Age)
devtools::install(upgrade = FALSE, quick = TRUE)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
ClinicoPath::statsplot(
data = deneme,
dep = Age,
group = Smoker)
mytable <- table(deneme$Outcome, deneme$Smoker)
caret::confusionMatrix(mytable)
confusionMatrix(pred, truth)
confusionMatrix(xtab, prevalence = 0.25)
levels(deneme$Outcome)
mytable[1,2]
d <- "0"
mytable[d, "FALSE"]
mytable[[0]]
formula <- jmvcore::constructFormula(terms = c("A", "B", "C"))
jmvcore::constructFormula(terms = list("A", "B", "C"))
vars <- jmvcore::decomposeFormula(formula = formula)
vars <- jmvcore::decomposeTerms(vars)
vars <- unlist(vars)
formula <- as.formula(formula)
my_group <- "lvi"
jmvcore::composeTerm(my_group)
my_dep <- "age"
formula <- paste0('x = ', group, 'y = ', dep)
myformula <- as.formula(formula)
myformula <- glueformula::gf({my_group}, {my_dep})
myformula <- glue::glue( 'x = ' , {my_group}, ', y = ' , {my_dep})
myformula <- jmvcore::composeTerm(myformula)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
library(survival)
km_fit <- survfit(Surv(OverallTime, Outcome) ~ LVI, data = deneme)
library(dplyr)
km_fit_median_df <- summary(km_fit)
km_fit_median_df <- as.data.frame(km_fit_median_df$table) %>%
janitor::clean_names(dat = ., case = "snake") %>%
tibble::rownames_to_column(.data = ., var = "LVI")
library(dplyr)
library(survival)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
myoveralltime <- deneme$OverallTime
myoutcome <- deneme$Outcome
myexplanatory <- deneme$LVI
class(myoveralltime)
class(myoutcome)
typeof(myexplanatory)
is.ordered(myexplanatory)
formula2 <- jmvcore::constructFormula(terms = "myexplanatory")
# formula2 <- jmvcore::decomposeFormula(formula = formula2)
# formula2 <- paste("", formula2)
# formula2 <- as.formula(formula2)
formula2 <- jmvcore::composeTerm(formula2)
formulaL <- jmvcore::constructFormula(terms = "myoveralltime")
# formulaL <- jmvcore::decomposeFormula(formula = formulaL)
formulaR <- jmvcore::constructFormula(terms = "myoutcome")
# formulaR <- jmvcore::decomposeFormula(formula = formulaR)
formula <- paste("Surv(", formulaL, ",", formulaR, ")")
# formula <- jmvcore::composeTerm(formula)
# formula <- as.formula(formula)
# jmvcore::constructFormula(terms = c(formula, formula2))
deneme %>%
finalfit::finalfit(formula, formula2) -> tUni
tUni
library(dplyr)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
results <- deneme %>%
ggstatsplot::ggbetweenstats(LVI, Age)
results
mydep <- deneme$Age
mygroup <- deneme$LVI
mygroup <- jmvcore::constructFormula(terms = "mygroup")
mygroup <- jmvcore::composeTerm(mygroup)
mydep <- jmvcore::constructFormula(terms = "mydep")
mydep <- jmvcore::composeTerm(mydep)
# not working
# eval(mygroup)
# rlang::eval_tidy(mygroup)
# !!mygroup
# {{mygroup}}
# sym(mygroup)
# quote(mygroup)
# enexpr(mygroup)
mygroup <- jmvcore::constructFormula(terms = "mygroup")
mydep <- jmvcore::constructFormula(terms = "mydep")
formula1 <- paste(mydep)
formula1 <- jmvcore::composeTerm(formula1)
mygroup <- paste(mygroup)
mygroup <- jmvcore::composeTerm(mygroup)
mydep <- deneme$Age
mygroup <- deneme$LVI
mydep <- jmvcore::resolveQuo(jmvcore::enquo(mydep))
mygroup <- jmvcore::resolveQuo(jmvcore::enquo(mygroup))
mydata2 <- data.frame(mygroup=mygroup, mydep=mydep)
results <- mydata2 %>%
ggstatsplot::ggbetweenstats(
x = mygroup, y = mydep )
results
myformula <- glue::glue('x = ', {mygroup}, ', y = ' , {mydep})
myformula <- jmvcore::composeTerm(myformula)
myformula <- as.formula(myformula)
mydep2 <- quote(mydep)
mygroup2 <- quote(mygroup)
results <- deneme %>%
ggstatsplot::ggbetweenstats(!!mygroup2, !!mydep2)
results
formula <- jmvcore::constructFormula(terms = c("myoveralltime", "myoutcome"))
vars <- jmvcore::decomposeFormula(formula = formula)
explanatory <- jmvcore::constructFormula(terms = c("explanatory"))
explanatory <- jmvcore::decomposeFormula(formula = explanatory)
explanatory <- unlist(explanatory)
myformula <- paste("Surv(", vars[1], ", ", vars[2], ")")
deneme %>%
finalfit::finalfit(myformula, explanatory) -> tUni
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
table3 <-
tangram::html5(
tangram::tangram(
"Death ~ LVI + PNI + Age", deneme),
fragment=TRUE,
# style = "hmisc",
style = "nejm",
# inline="nejm.css",
caption = "HTML5 Table",
id="tbl3")
table3
mydep <- deneme$Age
mygroup <- deneme$Death
formulaR <- jmvcore::constructFormula(terms = c("LVI", "PNI", "Age"))
formulaL <- jmvcore::constructFormula(terms = "Death")
formula <- paste(formulaL, '~', formulaR)
# formula <- as.formula(formula)
sty <- jmvcore::composeTerm(components = "nejm")
gr <- jmvcore::composeTerm(components = "Death")
table <- tangram::html5(
tangram::tangram(formula, deneme
),
fragment=TRUE,
# style = "hmisc",
# style = "nejm",
style = sty,
# inline="nejm.css",
caption = paste0("HTML5 Table ", gr),
id="tbl4")
table
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
mydata <- deneme
formula2 <- jmvcore::constructFormula(terms = c("LVI", "PNI", "Age"))
formulaR <- jmvcore::constructFormula(terms = "Death")
formulaR <- jmvcore::toNumeric(formulaR)
plot <-
finalfit::or_plot(
.data = mydata,
dependent = formulaR,
explanatory = formula2,
remove_ref = FALSE,
table_text_size = 4,
title_text_size = 14,
random_effect = NULL,
factorlist = NULL,
glmfit = NULL,
confint_type = NULL,
breaks = NULL,
column_space = c(-0.5, 0, 0.5),
dependent_label = "Death",
prefix = "",
suffix = ": OR (95% CI, p-value)",
table_opts = NULL,
plot_opts = list(
ggplot2::xlab("OR, 95% CI"),
ggplot2::theme(
axis.title = ggplot2::element_text(size = 12)
)
)
)
# Other Codes
## arsenal
tab1 <- arsenal::tableby(~ Age + Sex, data = deneme)
results <- summary(tab1)
# results$object
# results$control
# results$totals
# results$hasStrata
# results$text
# results$pfootnote
# results$term.name
#
# tab1$Call
#
# tab1$control
tab1$tables # this is where results lie
## define survival time
mydata$int <- lubridate::interval(
lubridate::ymd(mydata$SurgeryDate),
lubridate::ymd(mydata$LastFollowUpDate)
)
mydata$OverallTime <- lubridate::time_length(mydata$int, "month")
mydata$OverallTime <- round(mydata$OverallTime, digits = 1)
## Multivariate Analysis Survival
library(finalfit)
library(survival)
explanatoryMultivariate <- explanatoryKM
dependentMultivariate <- dependentKM
mydata %>%
finalfit(dependentMultivariate, explanatoryMultivariate) -> tMultivariate
knitr::kable(tMultivariate, row.names=FALSE, align=c("l", "l", "r", "r", "r", "r"))
# Find arguments in yaml
list_of_yaml <- c(
list.files(path = "~/histopathRprojects/ClinicoPath-Jamovi--prep/jmv",
pattern = "\\.yaml$",
full.names = TRUE,
all.files = TRUE,
include.dirs = TRUE,
recursive = TRUE
)
)
text_of_yaml_yml <- purrr::map(
.x = list_of_yaml,
.f = readLines
)
text_of_yaml_yml <- as.vector(unlist(text_of_yaml_yml))
arglist <-
stringr::str_extract(
string = text_of_yaml_yml,
pattern =
"([[:alnum:]]*):"
)
arglist <- arglist[!is.na(arglist)]
arglist <- unique(arglist)
arglist <- gsub(pattern = ":", # remove some characters
replacement = "",
x = arglist)
arglist <- trimws(arglist) # remove whitespace
cat(arglist, sep = "\n")
#
# # tUni_df_descr <- paste0("When ",
# # tUni_df$dependent_surv_overall_time_outcome[1],
# # " is ",
# # tUni_df$x[2],
# # ", there is ",
# # tUni_df$hr_univariable[2],
# # " times risk than ",
# # "when ",
# # tUni_df$dependent_surv_overall_time_outcome[1],
# # " is ",
# # tUni_df$x[1],
# # "."
# # )
#
# # results5 <- tUni_df_descr
boot::melanoma
rio::export(x = boot::melanoma, file = "data/melanoma.csv")
survival::colon
rio::export(x = survival::colon, file = "data/colon.csv")
# BreastCancerData <- "https://archive.ics.uci.edu/ml/machine-learning-databases/breast-cancer-wisconsin/breast-cancer-wisconsin.data"
#
# BreastCancerNames <- "https://archive.ics.uci.edu/ml/machine-learning-databases/breast-cancer-wisconsin/breast-cancer-wisconsin.names"
#
# BreastCancerData <- read.csv(file = BreastCancerData, header = FALSE,
# col.names = c("id","CT", "UCSize", "UCShape", "MA", "SECS", "BN", "BC", "NN","M", "diagnosis") )
library(mlbench)
data("BreastCancer")
BreastCancer
rio::export(x = BreastCancer, file = "data/BreastCancer.csv")
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
# names(deneme)
mypairwise <- survminer::pairwise_survdiff(
formula = survival::Surv(OverallTime, Outcome) ~ TStage,
data = deneme,
p.adjust.method = "BH"
)
mypairwise2 <- as.data.frame(mypairwise[["p.value"]]) %>%
tibble::rownames_to_column()
mypairwise2 %>%
tidyr::pivot_longer(cols = -rowname) %>%
dplyr::filter(complete.cases(.)) %>%
dplyr::mutate(description =
glue::glue(
"The comparison between {rowname} and {name} has a p-value of {round(value, 2)}."
)
) %>%
dplyr::select(description) %>%
dplyr::pull() -> mypairwisedescription
mypairwisedescription <- unlist(mypairwisedescription)
mypairwisedescription <- c(
"In the pairwise comparison of",
mypairwisedescription)
# mydata <- self$data
# mydep <- self$data[[self$options$dep]]
# mygroup <- self$data[[self$options$group]]
#
#
# plotData <- data.frame(gr = mygroup, dp = jmvcore::toNumeric(mydep))
# plotData <- jmvcore::naOmit(plotData)
#
# image <- self$results$plot
#
# image$setState(plotData)
# self$results$text1$setContent(plotData)
# mydepType <- data.frame(vclass = class(mydep),
# vtypeof = typeof(mydep),
# vordered = is.ordered(mydep),
# vfactor = is.factor(mydep),
# vnumeric = is.numeric(mydep),
# vdouble = is.double(mydep),
# vcharacter = is.character(mydep),
# vdate = lubridate::is.Date(mydep),
# vdate2 = is.na.POSIXlt(mydep)
# )
# mygroupType <- class(mygroup)
# variableTypes <- list(mydepType, mygroupType)
# self$results$text1$setContent(variableTypes)
# plotData <- image$state
# https://indrajeetpatil.github.io/ggstatsplot/
# ggbetweenstats violin plots for comparisons between groups/conditions
# ggwithinstats violin plots for comparisons within groups/conditions
#
# ggdotplotstats dot plots/charts for distribution about labeled numeric variable
#
# ggbarstats bar charts for categorical data
#
# ggscatterstats scatterplots for correlations between two variables
# http://corybrunson.github.io/ggalluvial/
# plot <- ggplot(plotData, aes(x = gr,
# y = dp)) +
# geom_point()
# plot <- plotData %>%
# ggstatsplot::ggbetweenstats(
# x = gr,
# y = dp
# )
library(readr)
BreastCancer <- read_csv("data/BreastCancer.csv")
View(BreastCancer)
mytarget <- "Class"
myvars <- c("Cl.thickness",
"Cell.size",
"Cell.shape",
"Marg.adhesion",
"Epith.c.size",
"Bare.nuclei",
"Bl.cromatin",
"Normal.nucleoli",
"Mitoses")
mydata <- BreastCancer %>%
select(mytarget, myvars)
formula <- jmvcore::constructFormula(terms = mytarget)
formula <- paste(formula, '~ .')
formula <- as.formula(formula)
# Create an FFTrees object from the data
FFTrees.fft <- FFTrees::FFTrees(
formula = formula,
data = mydata
)
# Plot the best tree applied to the test data
plot2 <- plot(FFTrees.fft,
data = mydata
# ,
# main = "Heart Disease",
# decision.labels = c("Healthy", "Disease")
)
devtools::install(upgrade = FALSE, quick = TRUE)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
ClinicoPath::statsplotbetween(
data = deneme,
dep = LVI,
group = PNI)
myirr <- data.frame(
Rater1 = c(0L,1L,1L,0L,0L,0L,1L,1L,1L,0L,1L,
1L,1L,1L,1L,0L,NA,1L,1L,0L,0L,1L,1L,1L,1L,1L,0L,
1L,1L,1L,1L,0L,1L,1L,1L,1L,1L,0L,0L,1L,1L,1L,
1L,1L,0L,1L,1L,1L,0L,0L,1L,1L,1L,0L,1L,1L,1L,0L,
1L,1L,0L,1L,0L,1L,1L,0L,0L,1L,0L,1L,1L,1L,0L,0L,
0L,0L,1L,1L,1L,0L,0L,1L,1L,1L,1L,0L,0L,0L,1L,0L,
0L,1L,1L,0L,1L,1L,0L,1L,1L,0L,1L,1L,0L,1L,1L,
0L,1L,1L,1L,0L,1L,1L,1L,0L,1L,1L,0L,0L,1L,0L,1L,
1L,1L,0L,1L,1L,1L,1L,1L,1L,1L,1L,0L,1L,1L,1L,1L,
1L,1L,1L,1L,1L,1L,0L,1L,1L,1L,1L,1L,1L,1L,0L,0L,
1L,0L,1L,1L,1L,1L,1L,0L,0L,1L,1L,1L,1L,1L,0L,
0L,0L,1L,1L,0L,1L,1L,0L,1L,0L,1L,1L,1L,0L,1L,1L,
1L,1L,1L,1L,1L,1L,1L,1L,1L,1L,1L,1L,1L,1L,1L,1L,
0L,0L,1L,1L,1L,1L,0L,0L,1L,1L,0L,1L,1L,1L,0L,1L,
0L,1L,1L,1L,1L,0L,0L,0L,0L,1L,0L,1L,1L,1L,0L,
0L,1L,1L,1L,0L,1L,0L,0L,0L,1L,1L,1L,0L,1L,0L,0L,
0L,1L,1L),
Rater2 = c(0L,0L,0L,0L,0L,0L,0L,0L,0L,0L,0L,
0L,0L,0L,0L,0L,0L,0L,0L,0L,0L,0L,0L,1L,1L,1L,0L,
1L,1L,1L,1L,0L,1L,1L,1L,1L,1L,0L,0L,1L,1L,1L,
1L,1L,0L,1L,1L,1L,0L,0L,1L,1L,1L,0L,1L,1L,1L,0L,
1L,1L,0L,1L,0L,1L,1L,0L,0L,1L,0L,1L,1L,1L,0L,0L,
0L,0L,1L,1L,1L,0L,0L,1L,1L,1L,1L,0L,0L,0L,1L,0L,
0L,1L,1L,0L,1L,1L,0L,1L,1L,0L,1L,1L,0L,1L,1L,
0L,1L,1L,1L,0L,1L,1L,1L,0L,1L,1L,0L,0L,1L,0L,1L,
1L,1L,0L,1L,1L,1L,1L,1L,1L,1L,1L,0L,1L,1L,1L,1L,
1L,1L,1L,1L,1L,1L,0L,1L,1L,1L,1L,1L,1L,1L,0L,0L,
1L,0L,1L,1L,1L,1L,1L,0L,0L,1L,1L,1L,1L,1L,0L,
0L,0L,1L,1L,0L,1L,1L,0L,1L,0L,1L,1L,1L,0L,1L,1L,
1L,1L,1L,1L,1L,1L,1L,1L,1L,1L,1L,1L,1L,1L,1L,1L,
0L,0L,1L,1L,1L,1L,0L,0L,1L,1L,0L,1L,1L,1L,1L,1L,
1L,1L,1L,1L,1L,1L,1L,1L,1L,1L,0L,1L,1L,1L,0L,
0L,1L,1L,1L,0L,1L,0L,0L,0L,1L,1L,1L,0L,1L,0L,0L,
0L,1L,1L)
)
myirr <- myirr %>%
dplyr::mutate(
RaterA = dplyr::case_when(
Rater1 == 0 ~ "Negative",
Rater1 == 1 ~ "Positive"
)
) %>%
dplyr::mutate(
RaterB = dplyr::case_when(
Rater2 == 0 ~ "Negative",
Rater2 == 1 ~ "Positive"
)
) %>%
dplyr::select(RaterA, RaterB) %>%
mutate(RaterA = as.factor(RaterA)) %>%
mutate(RaterB = as.factor(RaterB))
table <- myirr %$%
table(RaterA, RaterB)
mymatrix <- caret::confusionMatrix(table, positive = "Positive")
mymatrix
caret::sensitivity(table, positive = "Positive")
mymatrix2 <- caret::confusionMatrix(table, positive = "Positive", prevalence = 0.25)
mymatrix2
dat <- as.table(
matrix(c(670,202,74,640),
nrow = 2,
byrow = TRUE)
)
colnames(dat) <- c("Dis+","Dis-")
rownames(dat) <- c("Test+","Test-")
rval <- epiR::epi.tests(dat, conf.level = 0.95)
rval <- list(
dat,
rval,
print(rval),
summary(rval)
)
devtools::install(upgrade = FALSE, quick = TRUE)
library(dplyr)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
ratings <- deneme %>%
dplyr::select(LVI, PNI, Age, ID)
f <- unlist(lapply(ratings, class))
any(f == "numeric")
all(f == "numeric")
xtitle <- names(ratings)[1]
ytitle <- names(ratings)[2]
result <- table(ratings[,1], ratings[,2],
dnn = list(xtitle, ytitle))
table(ratings)
result1 <- irr::agree(ratings)
result2 <- irr::kappa2(ratings)
ClinicoPath::agreement(
data = deneme,
vars = c(LVI,PNI)
)
result2 <- irr::kappam.fleiss(
ratings = ratings,
exact = FALSE,
detail = TRUE)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
mytree <- vtree::vtree(deneme, "LVI PNI")
# write(mytree[["x"]][["diagram"]],
# file = here::here("/_tododata/trial1.gv"))
# DiagrammeR::grViz(diagram = here::here("/_tododata/trial1.gv"))
diagram <- mytree[["x"]][["diagram"]]
mytree2 <- DiagrammeR::grViz(diagram = diagram)
print(mytree2)
# Packages for Development
## rpkgtools
devtools::install_github("IndrajeetPatil/rpkgtools")
## available
Check if a package name is available to use https://docs.ropensci.org/available
https://github.com/r-lib/available
available::available("clinicopath")
available::available("lens2r")
## bench
High Precision Timing of R Expressions http://bench.r-lib.org/
https://github.com/r-lib/bench
## desc
Manipulate DESCRIPTION files
https://github.com/r-lib/desc
## pkgverse
pkgverse: Build a Meta-Package Universe
https://pkgverse.mikewk.com/
## pkgbuild
pkgbuild: Find Tools Needed to Build R Packages
https://github.com/r-lib/pkgbuild
## pkgload
pkgload: Simulate Package Installation and Attach
https://github.com/r-lib/pkgload
## rcmdcheck
rcmdcheck: Run 'R CMD check' from 'R' and Capture Results
https://github.com/r-lib/rcmdcheck
## remotes
## sessioninfo
Print Session Information
https://github.com/r-lib/sessioninfo
## "covr
## "exampletestr
## "covrpage",
## "gramr",
## "lintr",
## "goodpractice",
## "pkgdown",
## "usethis",
## "testthat",
## "spelling",
## "RTest",
https://towardsdatascience.com/rtest-pretty-testing-of-r-packages-50f50b135650
## "rhub",
## "roxygen2",
## "sinew",
## "styler",
## "vdiffr"
## "attachment (https://github.com/ThinkR-open/attachment)
## "covrpage (https://github.com/yonicd/covrpage)
## "defender (https://github.com/ropenscilabs/defender)
## "gramr (https://github.com/ropenscilabs/gramr)
## "packagemetrics (https://github.com/ropenscilabs/packagemetrics)
## "pRojects (https://github.com/lockedata/pRojects)
## "revdepcheck (https://github.com/r-lib/revdepcheck)
## "roxygen2Comment (https://github.com/csgillespie/roxygen2Comment)
## "roxygen2md (https://github.com/r-lib/roxygen2md)
## "testdown (https://github.com/ThinkR-open/testdown)
## "tic (https://github.com/ropenscilabs/tic)
# Table1 <- table(mydata[[testVariable]], mydata[[goldVariable]])
# Table1 <- mydata %>%
# janitor::tabyl(.data[[testVariable]], .data[[goldVariable]]) %>%
# janitor::adorn_totals(dat = ., where = c("row", "col")) %>%
# janitor::adorn_percentages(dat = ., denominator = "row") %>%
# janitor::adorn_percentages(dat = ., denominator = "col") %>%
# janitor::adorn_pct_formatting(dat = ., rounding = "half up", digits = 1) %>%
# janitor::adorn_ns(dat = .) %>%
# janitor::adorn_title("combined")
# results1 <- Table1
# results1 <- summary(km_fit)$table
# km_fit_median_df <- summary(km_fit)
# km_fit_median_df <- as.data.frame(km_fit_median_df$table) %>%
# janitor::clean_names(dat = ., case = "snake") %>%
# tibble::rownames_to_column(.data = .)
# results1 <- tibble::as_tibble(results1,
# .name_repair = "minimal") %>%
# janitor::clean_names(dat = ., case = "snake") %>%
# tibble::rownames_to_column(.data = ., var = self$options$explanatory)
table2 <- matrix(c(80, 20, 30, 70), nrow = 2, ncol = 2, byrow = TRUE, dimnames = list(c("Positive", "Negative"), c("Positive","Negative")))
table3 <- as.table(table2)
names(attributes(table3)$dimnames) <- c("Test","Gold Standart")
caretresult <- caret::confusionMatrix(table3, mode = "everything")
table3 <- matrix(c(80L, 20L, 25L, 30L, 70L, 75L), nrow = 2, ncol = 3, byrow = TRUE)
# RVAideMemoire::chisq.multcomp() RVAideMemoire::fisher.multcomp()
result1 <- RVAideMemoire::chisq.multcomp(table3)
result1 <- result1[["p.value"]]
result1 <- as.data.frame(result1) %>%
tibble::rownames_to_column()
result1 <- result1 %>%
tidyr::pivot_longer(cols = -rowname) %>%
dplyr::filter(complete.cases(.))
myfun <- function(i,j) {
if(!is.na(result1[i,j])){
paste0(
dimnames(result1)[[1]][i],
" vs ",
dimnames(result1)[[2]][j],
" p= ",
result1[i,j])
}
}
for (i in 1:dim(result1)[1]) {
for (j in 1:dim(result1)[2]) {
des <- myfun(i,j)
if(!is.null(des)) print(des)
}
}
myfun1 <- function(i,j) {
if(!is.na(result1[i,j])){
dimnames(result1)[[1]][i]
}
}
for (i in 1:dim(result1)[1]) {
for (j in 1:dim(result1)[2]) {
des <- myfun1(i,j)
if(!is.null(des)) print(des)
}
}
myfun(3,3)
myfun(1,2)
dimnames(result1)[[1]][2]
RVAideMemoire::fisher.multcomp(table3)
# rmngb::pairwise.chisq.test(x, ...) rmngb::pairwise.fisher.test(x, ...)
library(rmngb)
x <- sample(1:2, 1e3, TRUE)
g <- sample(1:4, 1e3, TRUE)
result2 <- rmngb::pairwise.chisq.test(x, g)
tab <- table(g, x)
resultrmngb <- rmngb::pairwise.fisher.test(tab, p.adj = "bonf")
result2[["p.value"]]
resultrmngb[["p.value"]]
rmngb::pairwise.chisq.test(tab)
formula <- jmvcore::constructFormula(terms = self$options$vars)
formula <- paste('~', formula)
formula <- as.formula(formula)
table1 <- arsenal::tableby(formula, self$data,
total = TRUE,
digits = 1,
digits.count = 1
)
myarsenal <- summary(table1, text = "html")
myarsenal <- kableExtra::kable(myarsenal, format = "html",
digits = 1,
escape = TRUE) %>%
kableExtra::kable_styling(kable_input = .,
bootstrap_options = "striped",
full_width = F,
position = "left")
library(dplyr)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
varsName <- c("LVI", "PNI")
tablelist <- list()
for (i in 1:length(varsName)) {
var <- varsName[i]
table <- deneme %>%
janitor::tabyl(dat = ., var) %>%
janitor::adorn_totals("row") %>%
janitor::adorn_pct_formatting(dat = .)
tablelist[[i]] <- table
}
tablelist
data <- self$data
vars <- self$options$vars
facs <- self$options$facs
target <- self$options$target
# data <- jmvcore::select(data, c(vars, facs, target))
if ( ! is.null(vars))
for (var in vars)
data[[var]] <- jmvcore::toNumeric(data[[var]])
if ( ! is.null(facs))
for (fac in facs)
data[[fac]] <- as.factor(data[[fac]])
data[[target]] <- as.factor(data[[target]])
data <- jmvcore::naOmit(data)
# TODO
# todo <- glue::glue(
# "This Module is still under development
# -
# -
# "
# )
# self$results$todo$setContent(todo)
# if (nrow(self$data) == 0)
# stop('Data contains no (complete) rows')
# if (is.null(self$options$vars) || is.null(self$options$target))
# return()
# prepare data for explore ----
# https://cran.r-project.org/web/packages/explore/vignettes/explore.html
# result1 <- iris %>% explore::explain_tree(target = Species)
#
# self$results$text1$setContent(result1)
# image <- self$results$plot
# image$setState(plotData)
# from https://forum.jamovi.org/viewtopic.php?f=2&t=1287
# library(caret)
# library(partykit)
# detach("package:partykit", unload=TRUE)
# library(party)
# Conditional Trees
# set.seed(3456)
# model <- train(
# yvar ~ .,
# data = df,
# method = 'ctree2',
# trControl = trainControl("cv", number = 10, classProbs = FALSE),
# tuneGrid = expand.grid(maxdepth = 3, mincriterion = 0.95)
# )
# plot(model$finalModel)
#
# t(sapply(unique(where(model$finalModel)), function(x) {
# n <- nodes(model$finalModel, x)[[1]]
# yvar <- df[as.logical(n$weights), "yvar"]
# cbind.data.frame("Node" = as.integer(x),
# psych::describe(yvar, quant=c(.25,.50,.75), skew = FALSE))
# }))
# data <- private$.cleanData()
# vars <- self$options$vars
# facs <- self$options$facs
# target <- self$options$target
# tree1 <- data %>%
# explore::explain_tree(target = .data[[target]])
# if (is.null(self$options$vars) || is.null(self$options$target))
# return()
# varsName <- self$options$vars
#
# facsName <- self$options$facs
#
# targetName <- self$options$target
#
# data <- jmvcore::select(self$data, c(varsName, facsName, targetName))
#
# data[[varsName]] <- jmvcore::toNumeric(data[[varsName]])
#
# for (fac in facsName)
# data[[facsName]] <- as.factor(data[[facsName]])
#
# data <- jmvcore::naOmit(data)
# tree1 <- data %>%
# explore::explain_tree(target = .data[[targetName]])
# plot <- iris %>% explore::explain_tree(target = Species)
# if (length(self$options$dep) + length(self$options$group) < 2)
# return()
# tree1 <- iris %>% explore::explain_tree(target = Species)
# iris$is_versicolor <- ifelse(iris$Species == "versicolor", 1, 0)
# tree2 <- iris %>%
# dplyr::select(-Species) %>%
# explore::explain_tree(target = is_versicolor)
# tree3 <- iris %>%
# explore::explain_tree(target = Sepal.Length)
library(magrittr)
# devtools::install(upgrade = FALSE, quick = TRUE)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
mydata <- deneme
varsName <- "Age"
# facsName <- c("LVI", "PNI")
targetName <- "Outcome"
mydata[[targetName]] <- as.factor(mydata[[targetName]])
mydata <- jmvcore::select(mydata, c(varsName,
# facsName,
targetName))
mydata <- jmvcore::naOmit(mydata)
explore::explain_tree(data = mydata,
target = targetName
)
mydata %>%
explore::explain_tree(target = .data[[targetName]])
iris %>% explore::explain_tree(target = Species)
BreastCancer %>%
dplyr::select(all_of(mytarget), all_of(myvars)) %>%
explore::explain_tree(target = .data[[mytarget]])
ClinicoPath::tree(
data = data,
vars = Age,
facs = vars(LVI, PNI),
target = Mortality)
mytarget <- "Class"
myvars <- c("Cl.thickness",
"Cell.size",
"Cell.shape",
"Marg.adhesion",
"Epith.c.size",
"Bare.nuclei",
"Bl.cromatin",
"Normal.nucleoli",
"Mitoses")
# mytarget <- jmvcore::composeTerms(mytarget)
# mytarget <- jmvcore::constructFormula(terms = mytarget)
# install.packages("easyalluvial")
library(magrittr)
# devtools::install(upgrade = FALSE, quick = TRUE)
deneme <- readxl::read_xlsx(path = here::here("_tododata", "histopathology-template2019-11-25.xlsx"))
mydata <- deneme
var1 <- "TStage"
var2 <- "Grade"
mydata <- jmvcore::select(df = mydata, columnNames = c(var1, var2))
mydata <- jmvcore::naOmit(mydata)
plot <-
easyalluvial::alluvial_wide( data = mydata
, max_variables = 5
, fill_by = 'first_variable'
, verbose = TRUE
)
plot %>%
easyalluvial::add_marginal_histograms(mydata)
imports <- c( attachment::att_from_rscripts(“./R”, recursive = TRUE) )
attachment::att_to_desc_from_is(path.d = “DESCRIPTION”, imports = imports, normalize = TRUE, add_remotes = TRUE)
so you’ve got a few options … but it’s worth pointing out that most of the time, the name of the jamovi function/analysis doesn’t really matter. unless you’re wanting people to be able to use the same functions from R, then no-one will ever use them/see them. (the only thing that matters is that the name of the analysis doesn’t change, so jamovi can match the analysis with analyses in old .omv files)
so assuming you want people to be able to use these same functions from R, and you want to rename, say, the flexplota function, you can use rename in the .a.yaml file. we use it for the anova:
https://github.com/jamovi/jmv/blob/master/jamovi/anova.a.yaml#L2-L3
this only renames the function used to call the analysis from R, and preserves the underlying ‘analysis name’ that jamovi depends on. in the case of our ANOVA, we decided the lowercase name conflicted with too many anova() functions in R. the only change this makes to your .h.R file is here:
https://github.com/jamovi/jmv/blob/master/R/anova.h.R#L527
another approach, if you don’t want to provide the analysis R functions at all, you can see export: false in the .a.yaml file.
https://github.com/jamovi/jmv/blob/master/jamovi/empty.a.yaml#L7
in the case of anova, this would completely remove the ANOVA() function from the .h.R file (jamovi doesn’t actually use that top level function itself, rather it constructs the anovaClass, anovaOptions, etc. objects directly) … some times people do this where they want to implement their own top level function, rather than relying on the automatically generated one. or sometimes they just don’t want people to use that function from R, and want them to use a different approach in R. (one usually wants to override the syntax generated for ‘syntax mode’ if taking this approach … i can walk you through that if that’s where you want to head). (edited) flatpak install flathub org.freedesktop.Sdk.Extension.gfortran-62
sudo apt install libcanberra-gtk-module libcanberra-gtk3-module
flatpak run --command=R --devel org.jamovi.jamovi
install.packages('node', repos='https://repo.jamovi.org')
install.packages('jmvtools', repos='https://repo.jamovi.org')
jmvtools::install(home='/app/bin/')Conclusion
This comprehensive guide covers the complete development lifecycle for ClinicoPath jamovi modules. For current development practices, focus on the sections covering:
-
Enhanced Development Workflow - Use the
configuration-driven approach with
_updateModules.R -
4-File Architecture - Master the
.b.R,.a.yaml,.r.yaml, and.u.yamlpatterns - Advanced Analysis Patterns - Implement complex analyses like stage migration and survival analysis
- Production Deployment - Follow the structured release and distribution process
For questions or contributions, please refer to the GitHub repository and community resources listed in the Reference Materials section.